

Ver Parte I: Azul de Metileno, guia básica
El excelente historial de seguridad del azul de metileno (MB) está bien establecido por un siglo de uso médico. Su papel recientemente explorado como potenciador mitocondrial y antioxidante de reciclaje es clave para su potencial como agente terapéutico. Con resultados alentadores en ensayos preclínicos de algunas de las enfermedades neurodegenerativas más discapacitantes, el MB debería investigarse con renovado entusiasmo en los próximos años. Si estos resultados se validan en futuros estudios en humanos, el MB podría demostrar ser un agente versátil que podría mejorar la salud de los pacientes que sufren la carga de trastornos neurodegenerativos y lesiones cerebrales, brindándoles a ellos y a sus seres queridos alivio y una mejor calidad de vida.
Resumen de los efectos del azul de metileno (MB) sobre los trastornos neurodegenerativos y las lesiones cerebrales y los mecanismos relacionados.
| Condición de interés | Acciones observadas de MB | Mecanismos de acción conocidos |
|---|---|---|
| Isquemia cerebral global | Neuroprotección [ 43 – 45 ]Neurológico [ 43 ]Promueve la función mitocondrial [ 43 ]Mejora la neuroprotección en combinación con hipotermia [ 46 , 47 ] | Oxida/inactiva la caspasa 3/6 [ 48 ]Inhibe la GC y la óxido nítrico sintasa [ 49 ]Estabiliza Hif-1α y fosforila Akt [ 44 ]Promueve la actividad IV compleja [ 43 ] |
| Ataque | Neuroprotección y disminución del tamaño del infarto y [ 50 – 53 ]Mejora neurológica [ 54 , 50 , 51 , 53 ]Preserva la estructura y función mitocondrial [ 54 , 51 ]Preserva la integridad de la barrera hematoencefálica [ 55 ]Mejora la neuroprotección en combinación con hiperoxia normobárica [ 56 ]Promueve la neurogénesis [ 54 ] | Regula positivamente las proteínas relacionadas con la mitofagia y la autofagia [ 51 ]Regula positivamente Akt y GSK-3β [ 55 ]Aumenta la regulación de la expresión y actividad del complejo IV [ 54 ]Disminuye la liberación de factores proinflamatorios [ 55 , 54 ] |
| Enfermedad de Alzheimer | Función cognitiva mejorada [ 57 – 62 ]Neuroprotección [ 57 ]Función mitocondrial preservada [ 57 , 30 , 63 ]Disminución de los niveles de Aβ [ 60 , 64 ]Disminución de la carga de tau [ 58 , 62 , 65 , 66 ]Aumento del flujo sanguíneo cerebral [ 59 ] | Inhibe la formación de Aβ oligomérico [ 67 ]Aumenta la depuración proteolítica de Aβ [ 64 ]Disminuye la actividad de la β-secretasa [ 60 ]Regula positivamente la actividad del complejo IV [ 30 , 63 ]Síntesis de hemo regulada positivamente [ 63 ]Disminución del estrés oxidativo [ 57 ]Previene la unión de tau-tau [ 66 ]Formación inhibida del filamento tau en el dominio de unión de microtúbulos [ 68 ]Residuos de cisteína tau oxidados [ 69 ] |
| Enfermedad de Parkinson | Mejoría neurológica [ 5 ]Mejora la función mitocondrial [ 5 ] | Transferencia alternativa de electrones mitocondriales [ 5 ] |
| Lesión cerebral traumática | Neuroprotección [ 70 – 72 ]Disminuye el tamaño del infarto [ 70 – 72 ]Disminuye el edema [ 73 ]Mejora la función neurológica [ 71 , 73 , 72 ] | Reduce la activación glial [ 70 ]Disminuye la liberación de factores proinflamatorios [ 73 ]Promueve la inducción de la autofagia [ 70 ] |
El azul de metileno, que es una molécula similar a la quinona, no es cualquier otro suplemento; es un agente poderoso que puede influir de forma importante en el metabolismo celular. Debido a que acepta y dona electrones, el azul de metileno mejora la función mitocondrial, ya que aborda problemas que a menudo pasan desapercibidos en la medicina convencional, como el estrés reductivo.
El estudio a continuación de esta introducción explica los beneficios multifacéticos de este compuesto. Pero antes de comenzar conviente considerar la advertencia del siguiente video reciente, publicado despúes de este estudio, que informa sobre un posible, aún por investigar efecto adverso (en ingles, Colabore con nosotros para que podamos subtitular este video)
ADVERTENCIA: La gastroenteróloga Dra. Sabine Hazan advierte que sus pacientes que usaron azul de metileno no pudieron aumentar su poblacion de bifidobacteria usando su protocolo cuando otros pacientes que no usaban azul de metileno, si lo lograron, ella se pregunta que puede estar sucediendo entre el azul de metileno y el microbioma de los primeros, este es un estudio que se debe realizar… Colabore con nosotros para que podamos subtitular este video https://x.com/i/status/1944503124833759569
El azul de metileno y la cadena de transporte de electrones
El azul de metileno tiene la habilidad de integrarse a la perfección en la cadena de transporte de electrones (ETC, por sus siglas en inglés), que influye en la generación de energía en las células. A diferencia de los antioxidantes tradicionales que donan o aceptan electrones, y que después requieren excretarse, el azul de metileno posee la capacidad única de alternar entre sus formas oxidada y reducida de manera indefinida.
Este proceso continuo de transferencia de electrones garantiza una mejora constante de la eficiencia de las mitocondrias, que es clave para la producción de energía y la salud celular general. Georgi enfatizó que el azul de metileno actúa como un oxidante de emergencia, ya que interviene para aceptar electrones incluso cuando los cofactores esenciales, como el dinucleótido de nicotinamida y la adenina (NAD+), son deficientes.
Esto hace que el azul de metileno pueda resolver problemas metabólicos asociados con la acumulación de electrones y el estrés reductivo. Debido a que mantiene el flujo de electrones dentro de la ETC, el azul de metileno evita el estancamiento que conduce a la disfunción celular y a diversos problemas de salud.

Este documento contiene la suficiente evidencia cientifica (más de 150 referencias) para que las madres puedan presentar a sus médicos y abogados y lograr exenciones para prevenir ser dañadas con vacunas o inyecciones génicas, que no tienen los suficientes estudios de seguridad como corresponde. Tambien sirve para educar a los médicos sin pensamiento crítico. descargar libro, click aqui
El azul de metileno para una mejor salud del cerebro
El potencial terapéutico del azul de metileno es muy amplio y variado, y abarca una gran variedad de afecciones neurológicas y psicológicas. Georgi compartió información sobre varios estudios en los que el azul de metileno, incluso en dosis bajas de 15 a 50 miligramos (mg), demostró beneficios significativos en el tratamiento de la depresión y la psicosis resistente al tratamiento.
Estos hallazgos son innovadores y sugieren que el azul de metileno mejora la función cognitiva y estabiliza el estado de ánimo, ya que mejora el rendimiento de las mitocondrias y reduce el estrés oxidativo en el cerebro. El azul de metileno favorece los beneficios de la niacinamida (vitamina B3) en la salud del cerebro y el metabolismo. Además, en términos de enfermedades neurodegenerativas, el azul de metileno ha demostrado ser muy prometedor.
Una versión modificada del azul de metileno, desarrollada por una empresa en Reino Unido, se patentó para el tratamiento del alzhéimer. Según Georgi, los ensayos clínicos presentaron una sorprendente reversión del 80 % de los síntomas del alzhéimer en los participantes, lo que destaca la capacidad del azul de metileno para detener y revertir el deterioro cognitivo.
Estas aplicaciones destacan la influencia del compuesto en el mejoramiento de la salud del cerebro, ya que garantizan una producción eficiente de energía y mitiga los efectos dañinos del estrés oxidativo. Una forma estabilizada de azul de metileno, conocida como hidrometiltionina (LMTM), también resulta prometedora para el tratamiento de la enfermedad de Alzheimer leve a moderada.
A diferencia del azul de metileno tradicional, el LMTM es una sal de dihidromesilato estabilizada, la cual ofrece propiedades farmacocinéticas mejoradas, incluyendo una mejor absorción en el cerebro y una vida media más larga en humanos. El estudio involucró a 1162 pacientes en dos ensayos de fase III, y demostró una actividad dependiente de la concentración de LMTM en el deterioro cognitivo y la atrofia cerebral.
En particular, la dosis terapéutica óptima se situó en torno a 16 mg al día, lo que maximizó los beneficios cognitivos sin los rendimientos decrecientes que se observaron con dosis mayores de 150 a 250 mg por día. Este efecto meseta resaltó que más allá de cierta concentración no se observaron beneficios adicionales, lo que coincidió con los hallazgos del estudio de que las dosis más altas no aportaron otras ventajas.
Además, el LMTM demostró beneficios significativos tanto por sí solo como en combinación con los tratamientos existentes para el alzhéimer. Los pacientes que recibieron LMTM mostraron un menor deterioro cognitivo y una atrofia cerebral más lenta, en comparación con aquellos con niveles de plasma más bajos. Esto sugiere que incluso en dosis más bajas y manejables, el LMTM retarda de manera eficaz la progresión del alzhéimer, ya que mejora la función mitocondrial.
Los beneficios importantes del azul de metileno en caso de shock séptico
Para ampliar las aplicaciones terapéuticas del azul de metileno, una revisión sistemática y metanálisis publicados en la revista Critical Care Explorations evaluaron la efectividad y seguridad del azul de metileno en pacientes con shock séptico, que es una afección con altas tasas de mortalidad.
El análisis incluyó seis ensayos controlados aleatorios con 302 pacientes, y buscó determinar si la administración de azul de metileno podría mejorar los resultados en comparación con el placebo o la atención habitual.
Los hallazgos sugieren que el azul de metileno puede reducir de forma significativa la mortalidad a corto plazo, acortar la duración del uso de vasopresores en un periodo de 31 horas y disminuir la estancia hospitalaria en un aproximado de dos días.
Además, el azul de metileno se asoció con un incremento en la presión arterial media a las seis horas después de administrarse. Cabe destacar que el estudio no encontró un aumento en los efectos adversos.
El azul de metileno inhibe la óxido nítrico sintetasa endotelial e inducible, lo que contrarresta la vasodilatación profunda que caracteriza al shock séptico. Al restaurar el tono vascular, el azul de metileno ayuda a mantener la perfusión y oxigenación adecuada de los órganos, que son necesarias para que el paciente sobreviva.

Este compendio de estudios de expertos, contiene la suficiente evidencia para que los padres puedan presentar a sus médicos y abogados y prevenir que su hijos sean intoxicados con vacunas o inyecciones génicas que no tienen los suficientes estudios de seguridad como corresponde. Tambien sirve para educar a los médicos sin pensamiento crítico. Click aqui para descargar este compendio
El azul de metileno para tratar el cáncer y combatir los tumores de ovario
Las investigaciones también exploraron el azul de metileno como un tratamiento para el cáncer de ovario, en particular para los casos resistentes a las quimioterapias convencionales. Un estudio publicado en la revista Cancers (Basel) utilizó en unos ratones un modelo de tumor de cáncer de ovario resistente al carboplatino, para así evaluar el impacto del azul de metileno en el crecimiento del tumor.
Los hallazgos revelaron una reducción significativa in vivo de la proliferación tumoral entre los ratones tratados con azul de metileno en comparación con los que recibieron únicamente carboplatino o que no tuvieron ningún tratamiento. En concreto, el azul de metileno demostró una supresión tumoral superior, lo que destacó su efectividad contra los tumores ováricos quimiorresistentes.
Otros análisis in vitro permitieron comprender mejor los mecanismos subyacentes a los efectos anticancerígenos del azul de metileno. El estudio investigó el impacto del azul de metileno en la energía mitocondrial en líneas celulares cancerosas y normales. El azul de metileno alteró la tasa de consumo de oxígeno y el potencial de la membrana mitocondrial dentro de las células de cáncer de ovario, lo que sugiere una mejor respiración mitocondrial e inducción de apoptosis.
Por el contrario, las células normales mostraron una respuesta muy diferente, con cambios menos pronunciados en la función mitocondrial, lo que indica una orientación selectiva del azul de metileno hacia las mitocondrias de las células cancerosas.
Se investigó la combinación de azul de metileno con una mezcla de ácido lipoico e hidroxicitrato y carboplatino para estudiar los efectos sinérgicos. Aunque la terapia combinada mostró una mejora leve en la respuesta tumoral en comparación con el azul de metileno por sí solo, la diferencia no fue significativa desde un punto de vista estadístico. Es importante destacar que las terapias metabólicas no indujeron toxicidad ni pérdida de peso en los ratones tratados, lo que resalta el buen perfil de seguridad de los tratamientos a base de azul de metileno.
El azul de metileno ofrece un enfoque novedoso que podría mejorar los resultados del tratamiento para los pacientes con opciones limitadas, ya que actúa sobre la función mitocondrial alterada e induce la apoptosis en células cancerosas quimiorresistentes. La respuesta diferencial entre las células cancerosas y las normales también sugiere que el azul de metileno ataca de forma selectiva el metabolismo tumoral, lo que minimiza el daño a los tejidos sanos.
El azul de metileno en situaciones de emergencia, como los infartos
Además de sus beneficios crónicos para la salud, el azul de metileno es muy valioso en las emergencias médicas agudas. Es efectivo para tratar problemas como el envenenamiento por cianuro y monóxido de carbono. En estos casos, el azul de metileno actúa rápido para restablecer la respiración celular, ya que acepta electrones y facilita el uso del oxígeno, lo que revierte los efectos tóxicos de estos venenos.
También le recomiendo que siempre tenga azul de metileno en su hogar, ya que es de gran ayuda en caso de infarto. Aunque la muerte súbita suele ser el resultado más común de la enfermedad cardíaca, las personas que sobreviven se enfrentan al daño por reperfusión, que es cuando se produce una mayor disfunción y muerte de las células después de restaurar el flujo sanguíneo.
La administración de azul de metileno mitiga en gran medida el daño tisular; sin embargo, es importante utilizar la dosis adecuada para evitar una sobredosis. Administre el azul de metileno a los pocos minutos del evento cardíaco para cumplir con el lapso de tiempo crítico y lograr su eficacia.
En casos de derrame cerebral o ataque cardíaco, incluso una dosis única menor a 50 mg podría salvarle la vida. Este beneficio inmediato convierte al azul de metileno en una herramienta esencial en la medicina de emergencia, ya que ofrece un medio rápido y efectivo para contrarrestar las crisis metabólicas.
El azul de metileno tiene capacidad para estabilizar rápido la función metabólica proporciona una capa adicional de protección contra los trastornos metabólicos repentinos que podrían ser letales. El potencial del azul de metileno para actuar como antídoto universal en diversas situaciones de intoxicación destaca su importancia, tanto en entornos médicos como de emergencia.
Los beneficios antienvejecimiento del azul de metileno
Las propiedades antienvejecimiento del azul de metileno son otra cuestión que Georgi explicó. Los estudios han indicado que el azul de metileno revierte el envejecimiento de las células humanas, ya que mantiene la función adecuada de las mitocondrias y reduce el daño oxidativo, que son factores clave en el proceso de envejecimiento. Las dosis diarias que van desde 5 mg a 50 mg alcanzan la concentración necesaria para estos beneficios sin causar decoloración en la orina o los tejidos.
Además, cuando se combina con la terapia de luz roja, los efectos del azul de metileno se amplifican de forma significativa. Esta sinergia promueve el rejuvenecimiento celular y la longevidad, ya que mejora la función de las mitocondrias y reduce el estrés oxidativo, lo que combate los signos visibles del envejecimiento y apoya la salud celular general.
El azul de metileno se puede utilizar en una dilución similar al enjuague bucal, lo que ofrece beneficios antisépticos sin los efectos secundarios agresivos de los enjuagues bucales convencionales. Esta forma de aplicación no solo aprovecha los beneficios metabólicos del azul de metileno, sino que también lo integra en las rutinas diarias con el fin de mejorar la salud y la longevidad.
Además de los beneficios principales mencionados, el azul de metileno exhibe otras propiedades prometedoras que podrían mejorar en gran medida diversos aspectos de la salud y la medicina. Funciona como un poderoso inhibidor de la aromatasa en concentraciones submicromolares, lo que podría presentar implicaciones en el manejo de afecciones relacionadas con las hormonas.
Además, la habilidad del azul de metileno para mejorar el flujo de electrones dentro de la cadena de transporte de electrones lo convierte en un suplemento versátil para tratar una amplia gama de trastornos metabólicos. Georgi Dinkov, experto en el área de la salud metabólicatambién introdujo el concepto de «Methylene Blue Test of Health», en el que la dosis a partir de la cual la orina de una persona comienza a tornarse azul sirve como indicador de su salud metabólica.
Un umbral de dosis menor para esta coloración sugiere una mejor función metabólica, mientras que umbrales mayores podrían indicar problemas de salud subyacentes, como cáncer o diabetes, que se caracterizan por estados de reducción extrema en las células.
Este enfoque innovador ofrece un método sencillo pero efectivo para que las personas controlen su salud metabólica y tomen medidas proactivas para tratar cualquier problema. A medida que avancen las investigaciones, es probable que se amplíe el espectro completo de beneficios del azul de metileno, lo que lo posicionaría como un elemento fundamental en las estrategias de salud preventivas y terapéuticas.

La seguridad de las vacunas contra la hepatitis B que se administran a los recién nacidos no se ha probado en un solo ensayo clínico controlado aleatorio con placebo inerte como se manifiesta en los propios prospectos y tiene sobredosis de aluminio neurotóxico. Este compendio de estudios de expertos, contiene la suficiente evidencia para que los padres puedan presentar a sus médicos y abogados y prevenir que su hijos sean intoxicados con vacunas que no tienen los suficientes estudios de seguridad como corresponde. Tambien sirve para educar a los médicos sin pensamiento crítico. Descargar libro click aqui
Algunas consideraciones de seguridad y dosificación
Aunque los beneficios del azul de metileno son considerables, es muy importante la dosificación adecuada para evitar efectos adversos graves que podrían ocurrir con dosis elevadas, en particular el síndrome serotoninérgico, que es una condición fatal causada por niveles excesivos de serotonina en el cerebro.
El azul de metileno es un potente inhibidor de la monoaminooxidasa tipo A (MAO-A), que puede elevar de forma peligrosa los niveles de serotonina cuando se combina con inhibidores selectivos de la recaptación de serotonina (SSRI, por sus siglas en inglés) u otros medicamentos serotoninérgicos. Se recomienda mucha precaución si toma algún medicamento SSRI, ya que no creo que nadie se beneficie de ellos.
Además, en dosis mayores de 30 mg a 50 mg, el azul de metileno puede causar que la orina y la lengua se tornen azules de forma temporal. Aunque no es peligroso, este efecto podría parecerle alarmante. Las dosis elevadas también pueden afectar las lecturas del oxímetro de pulso, lo que da lugar a evaluaciones inexactas de los niveles de oxígeno en la sangre.
Las personas con insuficiencia renal grave deben consumir el azul de metileno con precaución y bajo una estrecha supervisión médica, ya que el deterioro de la función renal afecta la depuración del medicamento. Además, el azul de metileno está contraindicado en pacientes con deficiencia de glucosa 6-fosfato deshidrogenasa (G6PD) debido al riesgo de anemia hemolítica.
Los efectos secundarios comunes relacionados con el azul de metileno incluyen:
- el malestar gastrointestinal leve y transitorio,
- como las náuseas y la diarrea,
- reacciones alérgicas, que van desde erupciones cutáneas hasta anafilaxia que podría ser mortal.
- Pueden ocurrir efectos neurológicos, como dolores de cabeza y confusión.
Aunque son menos comunes, los efectos cardiovasculares podrían incluir un aumento en la presión arterial y palpitaciones. Además, el azul de metileno interactúa con diversos medicamentos, en especial los antidepresivos y los antipalúdicos, lo que altera su efectividad o provoca reacciones adversas.
Para mitigar estos riesgos, Georgi recomienda dosis diarias más bajas de azul de metileno, por lo general entre 5 mg y 15 mg, sobre todo para uso a largo plazo. Estas dosis son suficientes para aprovechar los beneficios metabólicos sin aumentar de manera significativa el riesgo de síndrome serotoninérgico. Además, Georgi señaló que si bien las dosis más elevadas (hasta de 50 mg) demostraron ser efectivas en ciertas aplicaciones terapéuticas, deben utilizarse con precaución y bajo supervisión profesional.
Si está pensando en tomar suplementos de azul de metileno, consulte a un profesional de la salud capacitado para adaptar la dosis a sus necesidades específicas y evitar interacciones perjudiciales con otros medicamentos.
| Dosis diaria total | Mañana | Noche | |
| Semana 1 | 10mg | 10 gotas (5mg) | 10 gotas (5mg) |
| Semana 2 | 20mg | 20 gotas (5mg) | 20 gotas (5mg) |
| Semana 3 | 30mg | 30 gotas (5mg) | 30 gotas (5mg) |
Fuente: Mark Sloan, La Guía Definitiva del Azul de Metileno Solución al 1%
La siguiente es sólo información educativa y no sustituye la supervisión médica. Aunque el MB tiene un amplio margen de seguridad a dosis bajas, puede interactuar con antidepresivos (riesgo de síndrome serotoninérgico) y con enfermedades como deficiencia de G6PD.
Dosis según Mark Sloan (solución al 1 %)
Mark Sloan, en The Ultimate Guide to Methylene Blue (2021), recomienda la siguiente posología para uso oral en solución al 1 % (es decir, 10 mg/mL):
1 gota ≈ 0.05 mL = 0.5 mg de MB puro
- Dosis baja terapéutica general:
0.5 mg/kg/día (dividido en 2 tomas).
En un adulto de 70 kg → 35 mg/día, equivalentes a unas 70 gotas de una solución al 1 %.
Sloan enfatiza iniciar con dosis de 1–5 mg (2–10 gotas) para observar tolerancia, y aumentar gradualmente. - Rango de uso habitual según objetivos funcionales: Propósito Dosis recomendada Mejora cognitiva / Energía 5–15 mg por día (10–30 gotas) Neuroprotección / Fatiga crónica 10–30 mg por día (20–60 gotas) Apoyo mitocondrial en patologías 0.5–1 mg/kg por día (35–70 mg para 70 kg, es decir 70–140 gotas)
⚠️ Sloan recomienda siempre disolver en agua (por ejemplo, 100 – 200 mL) y evitar coadministrar con SSRIs o SNRIs.
Referencia:
(1) Sloan M. The Ultimate Guide to Methylene Blue: Remarkable Hope for Depression, Alzheimer’s, and Aging. Carnelian, 2021.
Dosis recomendadas en otros estudios revisados
Varios estudios revisados clínicamente han empleado dosis orales (o equivalentes) muy variables según el propósito:
- Neuroprotección / Terapia mitocondrial baja intensidad:
0.5–4 mg/kg/día, divididos en 2–3 dosis.
(2) Rojas JC, Gonzalez-Lima F. Methylene blue and neurodegeneration: mechanisms of action and therapeutic possibilities. Prog Neurobiol. 2011;96(1):32–45. - Ensayos en Alzheimer y deterioro cognitivo:
60–300 mg/día por vía oral (en formulación TRx0237/tauRx).
(3) Gauthier S et al. Phase 3 trials of leuco-methylthioninium for Alzheimer’s disease. J Alzheimers Dis. 2016;54(3):813–822. - Uso como agente mitocondrial en humanos sanos:
Dosis de 0.5–2 mg/kg demostraron mejoras cognitivas y mitocondriales medibles.
(4) Gonzalez-Lima F, Barrett DW. Augmentation of cognitive brain functions with low doses of methylene blue. Behavioural Brain Res. 2014;273:191–199. - Tratamientos coadyuvantes en shock séptico (oral y IV combinadas):
1–2 mg/kg; aunque clásicamente IV, las dosis equivalentes orales se sitúan en el mismo rango.
(5) Evora PRB et al. Methylene blue in the treatment of shock: a 20-year clinical experience. Ann Intensive Care. 2021;11:79.
Conversión práctica (solución al 1 %)
- 1 mL = 10 mg
- 1 gota ≈ 0.05 mL = 0.5 mg
| Dosis | Equivalente gotas (solución 1%) |
|---|---|
| 5 mg | ≈10 gotas |
| 10 mg | ≈20 gotas |
| 30 mg | ≈60 gotas |
| 60 mg | ≈120 gotas |
Recomendaciones prácticas
- Tomar con alimento o agua abundante para evitar irritación gástrica.
- No usar junto a antidepresivos serotoninérgicos.
- No usar si hay deficiencia de G6PD o embarazo.
- Comenzar con microdosis (2–3 gotas) y aumentar gradualmente.
Tabla comparativa de dosis orales de azul de metileno (solución al 1 %)
Tabla comparativa práctica de dosis de azul de metileno (MB) por vía oral, basada en Mark Sloan y la literatura médica revisada, en una solución al 1 % (10 mg/mL; 1 gota ≈ 0.05 mL = 0.5 mg).
| Objetivo terapéutico | Dosis (mg/día) | Gotas aproximadas | Comentarios clínicos | Referencias |
|---|---|---|---|---|
| Inicio / titulación | 1–5 mg | 2–10 gotas | Dosis inicial segura para evaluar tolerancia; útil en “microdosing”. | (1) |
| Rendimiento cognitivo / fatiga mental | 5–15 mg | 10–30 gotas | Mejora la eficiencia mitocondrial y la memoria a corto plazo. | (1)(4) |
| Neuroprotección / Trastornos mitocondriales leves | 10–30 mg | 20–60 gotas | Actúa como “carrier” alternativo en la cadena respiratoria. Puede mejorar claridad mental y energía. | (1)(2)(4) |
| Condiciones neurológicas moderadas (p. ej., Alzheimer temprano) | 60–150 mg | 120–300 gotas | En ensayos clínicos como TRx0237 se muestran efectos sobre proteínas tau. | (3) |
| Procesos degenerativos avanzados | 150–300 mg | 300–600 gotas | Altas dosis; uso experimental supervisado. Riesgo de toxicidad (náusea, insomnio, coloración urinaria azul). | (3) |
| Shock vasoplégico (comparativo a vía IV) | 1–2 mg/kg | 140 mg ≈ 280 gotas para 70 kg | Administrado preferentemente IV; la dosis oral es teóricamente equivalente, aunque de absorción más lenta. | (5) |
| Antidoto metahemoglobinemia leve (oral alternativa) | 1 mg/kg | 70 mg ≈ 140 gotas | Emergencias solo con guía médica; oral es secundaria a IV. | (5) |
Observaciones relevantes
- El rango óptimo mitocondrial (según Sloan y Gonzalez-Lima) está entre 0.5–4 mg/kg/día, con una curva en U: dosis demasiado altas pierden eficacia.
- Sloan subraya que la acción más beneficiosa aparece a niveles séricos bajos y sostenidos, por lo que algunos optan por 2–3 microdosis diarias.
- El color en la orina es marcador de absorción, no de toxicidad.
- El azul de metileno puede potenciar el metabolismo de la glucosa y el consumo de oxígeno cerebral, actuando como “bypass” para complejos mitocondriales dañados.
- Evitar combinación con fármacos serotoninérgicos (antidepresivos), pues inhibe MAO-A.
Referencias de la tabla comparativa de dosis
- Sloan M. The Ultimate Guide to Methylene Blue: Remarkable Hope for Depression, Alzheimer’s, and Aging. Carnelian Publishing; 2021.
- Rojas JC, Gonzalez-Lima F. Methylene blue and neurodegeneration: mechanisms of action and therapeutic possibilities. Prog Neurobiol. 2011;96(1):32–45.
- Gauthier S, et al. Phase 3 trials of leuco-methylthioninium for Alzheimer’s disease. J Alzheimers Dis. 2016;54(3):813–822.
- Gonzalez-Lima F, Barrett DW. Augmentation of cognitive brain functions with low doses of methylene blue. Behav Brain Res. 2014;273:191–199.
- Evora PRB, et al. Methylene blue in the treatment of shock: a 20-year clinical experience. Ann Intensive Care. 2021;11:79.
Guia Azul de Metileno oral solución al 1 %
Guia practica , basada principalmente en Mark Sloan (2021) y estudios de Gonzalez‑Lima y otros autores revisados, adaptada para una solución oral de azul de metileno al 1 % (10 mg/mL; 1 gota ≈ 0.05 mL = 0.5 mg).
Datos básicos
- Concentración: 1 % = 10 mg/mL
- 1 gota ≈ 0.5 mg de Azul de Metileno puro (MB)
- Dosis objetivo general: 0.5 – 4 mg/kg/día (dependiendo del propósito)
- Disolver siempre en 100–200 mL de agua o jugo ácido.
- Evitar: antidepresivos serotoninérgicos (ISRS, ISRN), IMAO, embarazo, deficiencia de G6PD.
⚙️ ETAPAS DE ESCALADO (para adultos ≥ 60 kg)
| Día | Dosis total (mg) | Gotas totales | Distribución recomendada | Objetivo |
|---|---|---|---|---|
| 1 – 3 | 1 mg | 2 gotas | 1 gota × 2 al día | Prueba de tolerancia / alergia |
| 4 – 6 | 5 mg | 10 gotas | 5 gotas AM | Activación ligera / enfoque |
| 7 – 10 | 10 mg | 20 gotas | 10 gotas AM | Metabolismo y claridad mental |
| 11 – 15 | 20 mg | 40 gotas | 20 AM / 20 PM | Aumento mitocondrial |
| 16 – 21 | 30 mg | 60 gotas | 30 AM / 30 PM | Plenitud funcional / neuroprotección |
| Mantenimiento | 10–30 mg | 20–60 gotas | 1–2 tomas/día | Según necesidad o respuesta |
Nota: En personas sensibles puede mantenerse microdosificación de 2–5 mg (4–10 gotas) diaria continua.
Protocolo según Objetivo
1️⃣ Revitalización cognitiva / enfoque mental
- Dosis: 5–15 mg/día (10–30 gotas)
- Timing: mañana o antes de actividad mental intensa
- Duración: 3–5 días/semana (con 2 días de descanso)
2️⃣ Fatiga crónica / disfunción mitocondrial leve
- Dosis: 10–30 mg/día (20–60 gotas)
- Dividir: mitad mañana, mitad tarde
- Duración: 4–6 semanas seguidas, reevaluar energía y sueño
3️⃣ Neuroprotección o envejecimiento cerebral
- Dosis: 0.5–1 mg/kg/día (~35–70 mg = 70–140 gotas en adulto 70 kg)
- Dividir: 2 tomás (AM + PM)
- Duración: uso cíclico — 3 semanas ON / 1 OFF
4️⃣ Situaciones clínicas graves (uso médico supervisado)
- Ej.: enfermedades neurodegenerativas moderadas‑severas.
- Dosis: 60–150 mg/día (120–300 gotas)
- Siempre bajo supervisión médica debido al posible estrés oxidativo o interferencia farmacológica.
Observaciones Clave
- El pico neuroquímico ocurre 60–90 min tras la ingesta oral.
- En dosis altas puede volverse oxidante y generar insomnio o irritabilidad.
- El color azul intenso en la orina indica absorción correcta.
- Puede emplearse junto con antioxidantes suaves (vitamina C, CoQ10, niacinamida), no con polifenoles reductores muy fuertes.
Referencias (Guia)
- Sloan M. The Ultimate Guide to Methylene Blue: Remarkable Hope for Depression, Alzheimer’s, and Aging. Carnelian Publ; 2021.
- Gonzalez‑Lima F, Barrett DW. Augmentation of cognitive brain functions with low doses of methylene blue. Behav Brain Res. 2014;273:191‑199.
- Rojas JC, Gonzalez‑Lima F. Methylene blue and neurodegeneration: mechanisms of action and therapeutic possibilities. Prog Neurobiol. 2011;96(1):32‑45.
- Gauthier S et al. Phase 3 trials of leuco‑methylthioninium for Alzheimer’s disease. J Alzheimers Dis. 2016;54(3):813‑822.
- Evora PRB et al. Methylene blue in the treatment of shock: a 20‑year clinical experience. Ann Intensive Care. 2021;11:79.
Efectos adversos y contraindicaciones del azul de metileno
El MB es claramente prometedor como tratamiento terapéutico para muchas enfermedades, pero, como cualquier fármaco, conlleva una serie de limitaciones. Las propiedades vasomoduladoras del MB son útiles en dosis terapéuticas, pero conllevan riesgos en concentraciones más altas. Estos riesgos van desde el aumento de la presión arterial y la resistencia vascular hasta alteraciones de la función cardíaca y la ritmicidad [ 2 ]. Asimismo, el MB puede influir en las mediciones de oximetría de pulso, lo que dificulta un diagnóstico adecuado.
Otro efecto tóxico es la inducción de anemia en individuos con deficiencia de glucosa-6-fosfato deshidrogenasa. Los neonatos también son susceptibles a la anemia y muchos otros efectos adversos como respuesta al MB [ 2 ]. En el neonato, el MB puede inducir hiperbilirrubinemia, que se presenta como ictericia [ 181 ]. Esto es bastante desafortunado, ya que el tratamiento aceptado para la hiperbilirrubinemia es la fototerapia, que activa la actividad fotodinámica del MB. Esto genera especies de oxígeno excitadas, que causan un daño significativo a la epidermis [ 182 ]. Por lo tanto, en cualquier aplicación, el MB debe tratarse con el cuidado de cualquier otro fotosensibilizador. En el neonato, el tratamiento con MB también puede conducir, paradójicamente, a metahemoglobinemia [ 183 ].
El efecto adverso final, y el más relevante clínicamente en el paciente adulto, está relacionado con el uso de medicamentos psiquiátricos comunes, los inhibidores selectivos de la recaptación de serotonina (ISRS). El MB es un Inhibidor de la monoaminooxidasa (IMAO) potente que, junto con inhibidores selectivos de la recaptación de serotonina (ISRS), puede potenciar el síndrome serotoninérgico, una emergencia médica potencialmente mortal [ 184–186 ] . Considerando la ubicuidad de los pacientes que toman ISRS, es prudente realizar pruebas de detección de estos medicamentos antes de administrar MB. De todos modos, el hecho de que los riesgos del MB sean relativamente bajos y razonablemente predecibles es un buen augurio para la adaptación en la clínica.
Recomendaciones para el consumo del azul de metileno
Por lo general, se venden tres tipos de azul de metileno:
- de grado industrial,
- de grado químico (grado de laboratorio) y
- de grado farmacéutico.
El único que debe utilizar es la variedad de grado farmacéutico en forma sólida, en cápsulas o en tabletas. Disolverlo en agua reduce bastante su efectividad después de 48 a 72 horas.
El azul de metileno es una opción popular para el mantenimiento de acuarios debido a sus propiedades antifúngicas, antiparasitarias y de transporte de oxígeno. Se suele utilizar para aliviar el estrés de los peces, combatir infecciones fúngicas y eliminar parásitos externos, como la enfermedad de las manchas blancas. Sin embargo, el azul de metileno apto para acuarios suele contener contaminantes dañinos, incluyendo metales pesados, que representan graves riesgos para la salud de las mascotas acuáticas.
Para garantizar la seguridad y el bienestar de sus mascotas, no le recomiendo utilizar productos de azul de metileno diseñados para acuarios en cualquier aplicación relacionada con mascotas. En su lugar, escoja el azul de metileno de grado farmacéutico, el cual se somete a pruebas rigurosas para comprobar que está libre de impurezas dañinas.
Dosis común
5 mg de azul de metileno al día, seis días a la semana.
Es importante enfatizar que la forma adecuada y legal de utilizar el azul de metileno es mediante una receta de un médico calificado. Si está considerando utilizar azul de metileno para su salud, le recomiendo mucho que consulte con su médico para determinar si es adecuado para sus necesidades y circunstancias específicas.
https://cienciaysaludnatural.com/prueba-de-mutacion-mthfr/ Alrededor del 40 por ciento de las personas en los Estados Unidos portan o están afectadas por la mutación MTHFR. Es una enzima encargada de transformar el folato (vitamina B9) en su forma activa. Ver en https://www.bitchute.com/video/AcSXBNVS3aDJ
Azul de metileno, de la función mitocondrial a la neuroprotección
From Mitochondrial Function to Neuroprotection – An Emerging Role for Methylene doi: 10.1007/s12035-017-0712-2 – https://pmc-ncbi-nlm-nih-gov.translate.goog/articles/PMC5826781/
El azul de metileno (MB) es un fármaco bien establecido con una larga historia de uso, debido a su amplia gama de usos y su perfil mínimo de efectos secundarios. El MB se ha utilizado clásicamente para el tratamiento de la malaria, la metahemoglobinemia y la intoxicación por monóxido de carbono, así como como colorante histológico.
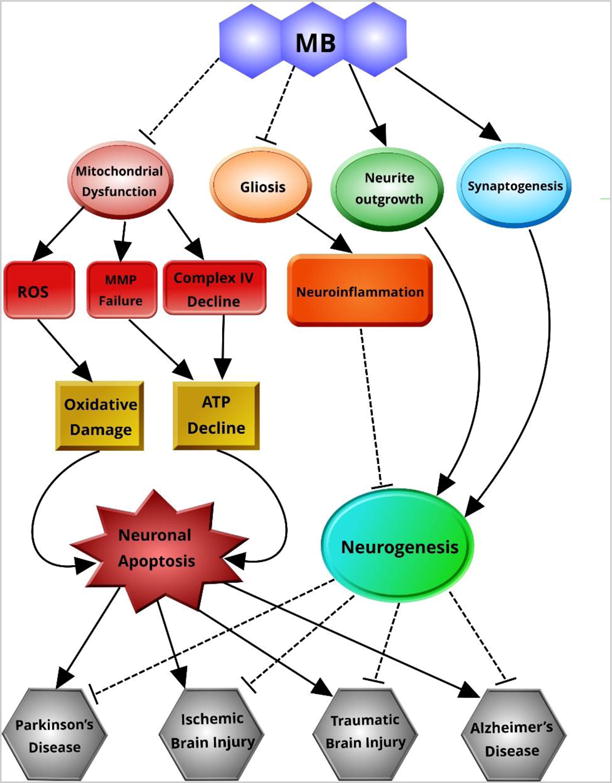
Sin embargo, su papel en las mitocondrias ha suscitado gran parte de su renovado interés en los últimos años. El azul de metileno (MB) promueve eficazmente la actividad mitocondrial al tiempo que mitiga el estrés oxidativo. Además de su efecto beneficioso sobre la protección mitocondrial, también se sabe que el azul de metileno (MB) tiene efectos sólidos en la mitigación de la neuroinflamación.
La disfunción mitocondrial se ha identificado como un fenómeno patológico aparentemente unificador en una amplia gama de trastornos neurodegenerativos, lo que posiciona al azul de metileno como una terapia prometedora.
En estudios in vitro e in vivo , el MB ha demostrado una eficacia impresionante para mitigar la neurodegeneración y los fenotipos conductuales que la acompañan en modelos animales para afecciones como accidente cerebrovascular, isquemia cerebral global, enfermedad de Alzheimer, enfermedad de Parkinson y lesión cerebral traumática.
Esta revisión resume trabajos recientes que establecen al MB como un candidato prometedor para la neuroprotección, con especial énfasis en la contribución de la función mitocondrial a la salud neuronal. Además, esta revisión examinará brevemente el vínculo entre el MB, la neurogénesis y la mejora de la cognición con respecto al deterioro cognitivo relacionado con la edad.
Este documento contiene la suficiente evidencia científica (más de 50) para que las madres puedan presentar a sus médicos y abogados y prevenir sus hijas e hijos sean dañados con vacunas que no tienen los suficientes estudios de seguridad como corresponde. Tambien sirve para educar a los médicos sin pensamiento crítico. No espere hasta último momento para estar protegida… descargar desde: https://cienciaysaludnatural.com/recursos

1. Introducción
La reutilización de fármacos establecidos da lugar a una traducción clínica mucho más rápidamente que un tratamiento novedoso, debido a un historial establecido de pruebas de seguridad exhaustivas. Esta estrategia es imperativa en campos de investigación de alto riesgo, como los trastornos neurodegenerativos, en los que las compañías farmacéuticas pueden ser reacias a invertir grandes sumas de dinero debido a un historial decepcionante de ensayos clínicos de alto nivel.
Afecciones como el accidente cerebrovascular, la enfermedad de Alzheimer (EA) y la enfermedad de Parkinson (EP) son devastadoras y están acompañadas de ramificaciones de por vida, lo que deja a los pacientes y a las familias con una duración y calidad de vida reducidas. Este impacto se ve agravado por la falta de opciones médicas que puedan ralentizar o revertir la progresión del trastorno. Estas afecciones necesitan urgentemente nuevas terapias y podrían beneficiarse enormemente del atractivo financiero y clínico de una nueva aplicación para un fármaco probado en el tiempo.
El azul de metileno (MB) es un ejemplo de un fármaco prometedor con potencial de reutilización. Sintetizado por primera vez en 1876 como tinte textil, el MB se investigó por sus aplicaciones medicinales ya en 1891 [ 1 ]. El MB se ha utilizado clásicamente en la clínica como un potente agente antipalúdico, tratamiento de la metahemoglobinemia y como agente de tinción médica [ 2 ].
La evidencia también ha demostrado que el MB es eficaz como terapia profiláctica contra el síndrome vasopléjico que se produce después de la cirugía de bypass de la arteria coronaria y en el choque séptico como resultado de la acción vasomoduladora del MB, que se basa en su inhibición de la guanilato ciclasa (GC) y la óxido nítrico sintasa [ 3 ].
Si bien la etiología y la progresión de los principales trastornos neurodegenerativos varían ampliamente, todos ellos tienen en común la disfunción mitocondrial. En afecciones neurodegenerativas como el accidente cerebrovascular, la enfermedad de Alzheimer, la enfermedad de Parkinson o la lesión cerebral traumática (LCT), la disfunción mitocondrial y el estrés oxidativo son clave para la naturaleza debilitante progresiva de la afección [ 4 ].
El deterioro de la cadena de transferencia de electrones mitocondrial, a través de un daño directo o una renovación insuficiente, conduce a déficits de energía y a la liberación de especies reactivas de oxígeno que pueden causar daños directos y posteriores y efectos nocivos [ 5 ].
Como tal, abordar y mejorar la salud mitocondrial es un objetivo principal para las terapias emergentes. El mecanismo mitocondrial del MB, así como su historial de seguridad establecido, lo sitúa como candidato para el tratamiento de estas afecciones devastadoras.
En esta revisión, se discutirán los mecanismos básicos de la disfunción mitocondrial en cada uno de los principales trastornos neurodegenerativos y se evaluarán los estudios recientes que examinan el efecto del MB en cada uno de ellos.
Se destacarán los estudios que se centran en el papel emergente del MB como antioxidante reciclador y transportador alternativo de electrones en la cadena de transferencia de electrones mitocondrial en el contexto de la neurodegeneración [ 5 ]. Cuando esté disponible, se comparará la terapia combinada con la monoterapia. También revisaremos brevemente la evidencia que sugiere un papel nootrópico para el MB.
2. Historia, mecanismos e indicaciones actuales del azul de metileno
El MB fue sintetizado originalmente por Hienrich Caro como tinte textil, pero rápidamente se descubrió que tenía un uso significativo en el campo de la medicina [ 1 ]. Los primeros estudios pioneros involucraron su uso como tinción médica, seguido poco después por su aplicación por Ehrlich y Guttman en el tratamiento de la malaria [ 6 ].
Este efecto lo convirtió en un fármaco importante en muchas campañas militares, aunque el efecto secundario de la orina azul no fue particularmente bien recibido. Si bien orinar de color azul era indeseable para los soldados, fue de gran utilidad en psiquiatría; incluido en la medicación, el MB proporcionó un indicador visiblemente aparente de cumplimiento [ 1 , 7 ].
Tal indicador fue invaluable en el tratamiento históricamente severo de los pacientes psiquiátricos con medicamentos tempranos que estaban plagados de efectos secundarios indeseables. A través de esta aplicación en psiquiatría, finalmente se descubrió que el MB solo tenía efectos antipsicóticos, lo que llevó al desarrollo de los tricíclicos.
El MB se utilizó como molécula piloto, marcando el comienzo de una nueva era de la psicofarmacología [ 1 , 8 ]. Se sabe que algunos de estos efectos se deben a su función como un potente inhibidor de la monoaminooxidasa (IMAO), pero trabajos recientes han implicado el papel de los mecanismos mitocondriales y la manipulación metabólica como la fuente de al menos algunos de los efectos psiquiátricos beneficiosos del fármaco [ 9 , 10 ]. En los últimos años, este trabajo ha extendido la aplicación psiquiátrica del MB al tratamiento de la depresión bipolar residual [ 11 ].
Estos resultados implican que el MB tiene biodisponibilidad en el cerebro, lo que está corroborado por estudios farmacocinéticos [ 12 ]. El MB se administra mejor por vía intravenosa, ya que la administración intravenosa proporciona concentraciones sanguíneas y AUC más altas que la administración oral, y tiene una vida media de aproximadamente 6,6 h.
Independientemente de la vía de administración, el MB se acumula en concentraciones significativas en varios tejidos, y la concentración de MB en el tejido cerebral es hasta diez veces mayor que los niveles séricos tan pronto como 1 hora después de la administración intravenosa [ 12 ].
La captación tisular es rápida, y se observa una acumulación sustancial en los órganos después de 3 minutos en los pulmones, el hígado, los riñones y el corazón [ 13 ]. La biodisponibilidad también está modulada por el estado de oxidación, ya que una versión estabilizada de la forma reducida de MB muestra una captación cerebral notablemente aumentada, lo que se ha tenido en cuenta en un ensayo clínico reciente que se analizará en una sección posterior [ 14 , 15 ].
Actualmente, los principales usos médicos de MB son metahemoglobinemia, síndrome vasopléjico, tinción quirúrgica y neurotoxicidad por ifosfamida [ 2 ].
La metahemoglobinemia es causada por una prevalencia de metahemoglobina (met-Hb), una forma de hemoglobina (Hb) en la que el centro ferroso del grupo hemo se oxida a un estado férrico. Esto puede ser hereditario o inducido por exposición a toxinas ambientales o ciertas drogas de abuso, como el nitrito de amilo [ 16 ].
La metahemoglobinemia se presenta como fatiga, dolores de cabeza, mareos y piel azulada y potencialmente puede conducir a convulsiones y muerte si no se trata. Una vez que MB se reduce a azul de leucoetileno (leucoMB) en los glóbulos rojos, leucoMB puede reducir met-Hb a Hb, reoxidando de nuevo a MB [ 1 , 17 ].
Esto se ilustró más famosamente en el caso de los Blue Fugates, una familia en la zona rural de Kentucky conocida por su piel azul como resultado de metahemoglobinemia hereditaria. El tratamiento alivió rápidamente el tono azul de su piel, para su gran alivio [ 18 ].
Si bien la metahemoglobinemia es uno de los usos más comunes del MB, sus otras aplicaciones son invaluables. Como tinción clínica, el MB brinda resultados sorprendentes en la detección de nervios y fístulas y se usa comúnmente en varios procedimientos, así como en tinción histológica [ 19 ].
El MB también se puede utilizar como terapia adjunta para la quimioterapia con ifosamida, un agente de quimioterapia común con efectos secundarios neurológicos perjudiciales a través del deterioro de la cadena de transferencia de electrones (CTE) mitocondrial.
Como transportador de electrones alternativo, el MB puede promover la función mitocondrial, limitando los efectos neurotóxicos del fármaco [ 20 ].
Finalmente, el síndrome vasopléjico es una afección potencialmente mortal que ocurre después de la derivación cardiopulmonar y se manifiesta como una presión arterial significativamente reducida, especialmente prevalente en el caso de pacientes con antecedentes de inhibidores de la enzima convertidora de angiotensina. La administración de MB puede interceder en esta afección a través de la inhibición de la guanilato ciclasa y la óxido nítrico sintasa, aumentando la presión arterial [ 21 ]. Debido a estas importantes y necesarias aplicaciones médicas, el MB es reconocido como uno de los medicamentos necesarios en un sistema básico de atención sanitaria [ 22 ].
El daño oxidativo, una causa y consecuencia de la disfunción mitocondrial, afecta principalmente al complejo IV así como al complejo I [ 24 ]. Este bloqueo también es eludido por el MB, ya que puede aumentar significativamente la actividad del complejo IV. La expresión de las subunidades del complejo IV se regula posteriormente al alza, tal vez a través de la inducción del factor respiratorio nuclear 1 ( Nrf1 ) , que se encontró elevado en ratones viejos tratados con MB [ 25–27 ].
3. Azul de metileno, disfunción mitocondrial y neurodegeneración
El cerebro depende notablemente del metabolismo oxidativo como fuente de energía, consumiendo el 20% de la glucosa del cuerpo y el 20% de su oxígeno en estado de reposo [ 31 ]. El mantenimiento del potencial de membrana en reposo, la generación de potenciales de acción y las acciones postsinápticas del glutamato comprenden la mayor parte de esta demanda de energía que está estrechamente relacionada con la actividad neuronal [ 32 , 33 ].
Considerando la sorprendente falta de fuentes de energía alternativas en el cerebro, la función mitocondrial adecuada es imperativa para la salud cerebral. Las mitocondrias disfuncionales, por el contrario, están implicadas en varias enfermedades neurodegenerativas, desempeñando un papel causal o contribuyente [ 4 ].
La función principal de la cadena de transporte de electrones mitocondrial es transferir electrones de alta energía desde sustratos energéticos derivados de los alimentos como el NADH al O 2 de manera gradual. Cada paso libera energía que se utiliza para transportar protones a través de la membrana mitocondrial interna, estableciendo el potencial transmembrana que impulsa la ATP sintasa, el rotor molecular que convierte el ADP en ATP [ 23 , 34 ].
El funcionamiento mitocondrial adecuado depende del paso secuencial de electrones a través de cada paso de la cadena de transporte de electrones. A medida que la cadena de transporte de electrones se ocupa al máximo, los transportadores de electrones comienzan a donar electrones al O 2 produciendo ROS perjudiciales [ 34 ].
Las proteínas son frecuentemente el objetivo del ataque de radicales libres. Las modificaciones a las proteínas por ROS/RNS incluyen, pero no se limitan a: oxidación, nitrosilación, acetilación y fosforilación [ 36 ]. Como las mitocondrias son la fuente de ROS y tienen varias metaloproteínas que pueden catalizar la formación de ROS como los radicales hidroxilo, son fuertemente atacadas por daño oxidativo [ 34 ]. Los componentes de la cadena de transporte de electrones mitocondrial son objetivos de daño oxidativo, lo que lleva a una degradación metabólica progresiva que contribuye a la neurodegeneración [ 37 ] ( Fig. 3 ).
La función mitocondrial también depende de la dinámica de fisión/fusión y la mitofagia, cuyos componentes esenciales pueden resultar dañados por daño nitroxidativo [ 38 ]. La dinámica de fisión/fusión está regulada por las GTPasas de la familia de la dinamina y sirve para mantener la función mitocondrial mediante el intercambio de componentes mitocondriales y la degradación de mitocondrias dañadas a través de la mitofagia [ 4 ].
El daño oxidativo a la proteína relacionada con la fusión OPA1 da como resultado la translocación al citosol seguida de fragmentación mitocondrial [ 39 ]. La fragmentación mitocondrial es frecuente en varios trastornos neurodegenerativos, como se analizará con más detalle en secciones posteriores de esta revisión [ 40 ]. El transporte mitocondrial está intrínsecamente vinculado a la dinámica de fisión/fusión y se ha demostrado que está alterado en las enfermedades neurodegenerativas [ 4 , 41 ].
Todos estos efectos perjudiciales se ven exacerbados por el daño del ADNm, que conduce a mutaciones dañinas que se distribuyen de forma heterogénea entre las mitocondrias dentro de una sola célula. Este daño impide la reparación mitocondrial adecuada al deshabilitar de forma preventiva las proteínas antes de que se traduzcan.
Esto garantiza que el daño a las mitocondrias sea duradero e incluso pueda propagarse durante los eventos de fisión/fusión [ 42 ]. Estas mutaciones se agravan aún más por la dinámica de fisión/fusión deteriorada y la mitofagia mal regulada [ 34 ]. Es importante destacar que todos estos daños pueden perpetuarse entre sí de manera progresiva, lo que conduce a una disfunción mitocondrial progresiva que contribuye al avance inexorable de la neurodegeneración [ 38 ].

El aluminio en las vacunas es neurotóxico y el calendario de vacunación infantil tiene sobredosis de aluminio. Los estudios de seguridad del aluminio tienen graves errores y este tema esta postergado desde hace décadas. Más de 100 referencias científicas de expertos para que Usted presente a su abogado o médico, para eximir a sus hijos de las vacunas. Descargar libro click aqui
3.1 Azul de metileno y lesión cerebral isquémica
Como se señaló anteriormente, el cerebro es extremadamente sensible a la privación de oxígeno y glucosa como resultado de sus altas demandas de energía [ 31 ]. La isquemia, o cese del flujo sanguíneo, al cerebro puede ser focal o global.
La isquemia cerebral global (ICG) es la pérdida completa del flujo sanguíneo cerebral (FSC) y ocurre durante un traumatismo como un paro cardíaco, asfixia, cirugía cardíaca y shock hipotensivo. El paro cardíaco requiere una intervención rápida; incluso con reanimación, suele ser fatal, con una tasa de mortalidad de más del 90%.
Para aquellos que sobreviven, les espera una serie de posibles discapacidades. Los sobrevivientes deben lidiar con déficits sensoriomotores, desregulación del estado de ánimo, pérdida de memoria y deterioro cognitivo que resultan de la muerte celular neuronal tardía en todo el cerebro, específicamente en el hipocampo [ 74 , 75 ].
La única intervención indicada actualmente para prevenir la muerte celular inducida por GCI es la hipotermia terapéutica (HT), que ralentiza el metabolismo oxidativo y potencialmente mitiga el estrés oxidativo inducido por la reperfusión y el descarrilamiento mitocondrial [ 76 , 77 ] [ 78 ].
La inducción temprana de HT es imperativa, ya que la HT ofrece rendimientos decrecientes a medida que aumenta la duración después de GCI. Además, la inducción de HT requiere un equipo especializado sustancial y capacitación del personal para prevenir efectos secundarios significativos [ 79 ].
Esto limita la implementación, lo que requiere investigación en terapias alternativas que se puedan implementar con mayor facilidad y accesibilidad.
La isquemia cerebral focal, o accidente cerebrovascular, ocurre casi cada 40 segundos en los Estados Unidos y mata o daña significativamente al 60% de los pacientes. El accidente cerebrovascular exige un alto costo emocional, médico y económico que cuesta casi $33 mil millones cada año en los EE. UU. [ 80 ].
Hay dos categorías principales de accidente cerebrovascular,
- isquémico
- o hemorrágico [ 80 ].
La mayoría de los accidentes cerebrovasculares son isquémicos, resultantes del bloqueo de una arteria cerebral, generalmente por aterotrombosis [ 80 ]. El núcleo central de la región isquémica sufre rápidamente una muerte celular predominantemente necrótica, mientras que la región de penumbra circundante sufre una falla energética progresiva, inflamación y muerte celular apoptótica tardía [ 81 ].
Actualmente, solo hay una intervención indicada en el tratamiento del ictus, la trombolisis con activador tisular del plasminógeno (tPA) [ 82–84 ]. Esto requiere un diagnóstico adecuado mediante técnicas de imágenes cerebrales, que consumen la ventana de tiempo de 4 horas en la que se puede salvar el núcleo del infarto [ 83 ] . Dado que el ictus a menudo no se identifica dentro de esta ventana de tiempo, se necesitan desesperadamente estrategias farmacológicas que puedan minimizar o prevenir la degeneración neuronal, especialmente aquellas que se pueden implementar de manera fácil y económica.
La isquemia cerebral, focal o global, se desarrolla con dos fases de lesión:
- isquemia y
- reperfusión.
Durante el inicio de la isquemia, la falla energética primero altera el mantenimiento del potencial de membrana neuronal. La despolarización posterior desencadena una liberación glutamatérgica excesiva que induce una rápida entrada de Ca 2+ .
El aumento brusco de los niveles intracelulares de Ca 2+ activa las fosfolipasas, calpaínas y catepsinas, aumenta la actividad de COX-2 y NOS e induce el cese de la síntesis de proteínas [ 85 , 86 ]. Mientras tanto, a medida que la respiración mitocondrial se detiene, la fuga de electrones genera ROS que dañan los componentes mitocondriales y celulares de las neuronas y las células vecinas.
El endotelio vascular responde a este daño oxidativo y la liberación local de factores inflamatorios permeabilizando y comprometiendo la integridad de la barrera hematoencefálica (BHE) [ 87 ].
Tras la reperfusión, el O2 y los sustratos energéticos vuelven a las mitocondrias ahora disfuncionales. La respiración se reanuda dentro de las mitocondrias defectuosas, liberando niveles cada vez mayores de ROS, lo que exacerba aún más el daño oxidativo.
La permeabilización de la BHE permite que los macrófagos circulantes se infiltren y perpetúen la inflamación local [ 4 ]. Mientras tanto, la microglia y los astrocitos responden a través de la activación glial, liberando en el medio extracelular sus propios factores inflamatorios [ 88 ].
Con el tiempo, este ciclo que se autoperpetúa finalmente se manifiesta como muerte celular apoptótica en la penumbra en la isquemia focal o en regiones particularmente sensibles, como el hipocampo, en la GCI [ 89 ]. Claramente, la salud mitocondrial es un contribuyente importante a la degeneración neuronal. Como tal, la disfunción mitocondrial se ha convertido en un objetivo atractivo para la neuroprotección contra el daño isquémico.
Se ha demostrado que el MB es neuroprotector en varios modelos de lesión isquémica, debido en parte a su capacidad antioxidante y su capacidad para facilitar la transferencia mitocondrial alternativa.
Dicho esto, gran parte del trabajo en el campo se ha centrado en la inhibición de la actividad de la NOS por parte del MB. Gran parte de este trabajo se ha realizado en modelos de lechones de paro cardíaco en el laboratorio de Wiklund. En 2007, demostraron que la administración de MB en el modelo de paro cardíaco de lechones podría aumentar significativamente la supervivencia durante 5 horas si se administraba 1 minuto después de la reanimación. Curiosamente, observaron que el MB atravesaba la barrera hematoencefálica y más tarde descubrieron que podía preservar la integridad de la BHE [ 90 , 45 ].
Este grupo descubrió más tarde que el tratamiento con MB aumentaba la circulación sistémica a través de la inhibición de la NOS y reducía las medidas de peroxidación lipídica e inflamación. Estos efectos protectores se observaron tanto en el tejido cerebral como en el cardíaco en el modelo porcino [ 45 ].
Los efectos neuroprotectores de la MB se amplificaron significativamente cuando se combinó con hipotermia tanto en cerdos como en roedores [ 46 , 47 ], tal vez debido al mantenimiento de la función mitocondrial adecuada mientras se desacelera el metabolismo e induce respuestas celulares hipotérmicas [ 91 ]. Es posible que la eficacia de la TH pueda mejorarse con la adición de un promotor de la función mitocondrial, especialmente uno que pueda mitigar el daño oxidativo. Sin embargo, se requiere más evidencia para llegar a una conclusión sólida sobre la perspectiva de la terapia combinada MB + TH.
Estudios de cultivos celulares de otros laboratorios han demostrado que algunos de los efectos neuroprotectores del MB están relacionados con la estabilización de Hif-1α y la fosforilación de la vía Akt [ 44 ]. Algunos de los efectos pueden deberse a la inhibición de la caspasa. El MB también puede oxidar el residuo de cisteína funcional tanto en la caspasa-3 como en la caspasa-6, impidiendo su acción proteolítica [ 48 ]. Este fenómeno también podría ser la base de la neuroprotección del MB y debería investigarse más a fondo.
Los mismos mecanismos neuroprotectores mitocondriales de MB demostrados en GCI también han mostrado eficacia en varios modelos animales de accidente cerebrovascular. MB podría aumentar el flujo sanguíneo cerebral en tejido hipoperfundido en un modelo de MCAO permanente en ratas al bloquear las acciones vasomoduladoras de NO a través de la inhibición de GC y NO sintasa, previniendo el desarrollo creciente de la penumbra [ 52 , 49 ].
El mecanismo mitocondrial subyacente de MB fue validado en modelos de accidente cerebrovascular, con mayor captación de O 2 , actividad del complejo IV y contenido de ATP [ 54 , 50 , 51 ]. Otros trabajos han demostrado repetidamente que el tamaño del infarto se puede controlar o reducir con MB, y que esta protección es proporcional a mejores resultados conductuales en modelos de roedores [ 44 , 53 ]. Los mecanismos por los cuales esto se logra son variados.
En un estudio antes mencionado, se demostró que MB logró rescatar la estructura mitocondrial y el potencial de membrana mitocondrial mientras preservaba la mitofagia [ 51 ].
Estudios similares han relacionado la inducción de autofagia por MB con la activación de mTOR y la fosforilación de AKT [ 44 , 92 ]. En particular, un trabajo reciente que aplicó MB a la hemorragia subaracnoidea, una forma de accidente cerebrovascular hemorrágico notablemente subrepresentada en la investigación de MB, mostró aumentos similares en la fosforilación de AKT y GSK-3β que fueron acompañados por una función neurológica mejorada y una reducción de la neuroinflamación [ 55 ].
En general, se encontró que MB inhibe las vías proapoptóticas y confiere neuroprotección contra la muerte celular en la región de penumbra, en algunos casos preservando estructuras celulares críticas como los pies terminales astrocíticos [ 93 , 50 ].
Como se mencionó anteriormente, el trabajo en nuestro laboratorio encontró que MB podría promover la neuroprotección al disminuir la activación de la caspasa y proteger el potencial de membrana mitocondrial.
Los resultados también se correlacionaron con un mejor desempeño conductual en el laberinto de Barnes, una prueba clásica de memoria espacial dependiente del hipocampo [ 43 ]. Nuestro trabajo posterior aplicó el tratamiento con MB a un modelo de accidente cerebrovascular fototrombótico de rata. Demostramos neuroprotección contra la muerte celular neuronal, así como la promoción de la neurogénesis inducida por accidente cerebrovascular. Estos resultados fueron paralelos a disminuciones en la inflamación microambiental [ 54 ]. Proponemos que esto es un resultado del papel de las mitocondrias en la neurogénesis, que se discutirá en mayor detalle en una sección posterior.
La terapia combinada se está explorando con gran interés, a pesar de la complejidad inherente del diseño del estudio. Un ejemplo novedoso de este tratamiento combinado de hiperoxia normobárica con MB. La terapia combinada disminuyó el volumen del infarto y produjo mejoras conductuales en un modelo de accidente cerebrovascular más allá de las de cada tratamiento por separado [ 56 ].
Nuestro trabajo encontró que la adición de MB a la hipotermia terapéutica logró rescatar la muerte celular neuronal y los resultados conductuales después de un GCI prolongado. Además, la terapia combinada redujo significativamente la activación glial, la inflamación y las vías apoptóticas de la caspasa 3.
Esto se produjo junto con la mejora de la disfunción mitocondrial. Todos estos efectos aumentaron notablemente en la terapia combinada en comparación con la monoterapia [ 46 ]. Por lo tanto, es probable que la terapia combinada sea la clave para maximizar los efectos beneficiosos de MB, especialmente considerando la amplia gama de alcance y variedad de lesión cerebral isquémica, accidente cerebrovascular en particular.
Si MB demuestra ser exitoso en el tratamiento de la lesión cerebral isquémica, focal o global, podría abrir la puerta a un tratamiento efectivo en diversos entornos de atención médica debido a su distribución generalizada en todo el mundo.

Los efectos secundarios de la vacuna contra el Sarampión, Rubeola y Paperas, SRP (MMR en EE.UU.) incluyen convulsiones, que ocurren en aproximadamente 1 de cada 640 niños vacunados, aproximadamente 5 veces más frecuentemente que las convulsiones por infección de sarampión, sepa como eximir a sus hijos de esta vacuna. Este compendio de estudios de expertos, contiene la suficiente evidencia para que los padres puedan presentar a sus médicos y abogados y prevenir que su hijos sean intoxicados con vacunas que no tienen los suficientes estudios de seguridad como corresponde. Tambien sirve para educar a los médicos sin pensamiento crítico. Descargar libro click aqui
Azul de metileno y enfermedad de Alzheimer
La enfermedad de Alzheimer, EA es un trastorno neurodegenerativo progresivo que afecta a 1 de cada 9 personas mayores de 65 años y que provoca:
- pérdida de memoria,
- deterioro cognitivo,
- discapacidad grave y muerte [ 70 ].
Actualmente, no existe un tratamiento eficaz para la EA que pueda detener o ralentizar significativamente el inexorable deterioro del paciente. En este sentido, esto hace que la EA sea única entre las principales causas de muerte en los EE. UU., ya que es una de las pocas causas principales de muerte con pocas medidas preventivas o terapéuticas [ 70 ].
Teniendo en cuenta el crecimiento de la comunidad de edad avanzada como resultado de una atención médica mejor y más accesible, la importancia de las opciones terapéuticas para controlar la EA solo se volverá más crítica con el tiempo. El desarrollo de medicamentos que puedan dirigirse a los mecanismos clave, o a múltiples mecanismos en el caso de MB, es imperativo.
Clásicamente, el principal enfoque de la investigación terapéutica de la EA se ha centrado en el Aβ. El MB ha comenzado a estudiarse en este sentido tan recientemente como en 2007. Los primeros trabajos descubrieron que el MB puede promover la fibrilación del Aβ, inhibiendo así la formación del Aβ oligomérico neurotóxico, aunque estudios in vitro posteriores fueron contradictorios [ 96 , 67 ]. El trabajo realizado en ratones transgénicos (3xTg-AD) ha descubierto que el MB apoyaba la depuración proteolítica del Aβ al aumentar la actividad de la quimotripsina y del proteosoma similar a la tripsina en el cerebro [ 64 ]. Se observó una disminución de la deposición de Aβ en el hipocampo y la corteza vecina en otro modelo de ratón transgénico (APP/PS1), y estas observaciones fueron apoyadas por una protección proporcional contra el deterioro cognitivo en tareas conductuales que miden la interacción social, el aprendizaje y la memoria, y la actividad exploratoria [ 61 ]. Se informaron resultados similares en el ratón transgénico PSAPP en el que se determinó que el mecanismo antiamiloidogénico estaba relacionado con la atenuación de la actividad y la expresión de la β-secretasa [ 60 ].
Disfunción mitocondrial en la aparición temprana de la patología de EA, presente antes de la deposición significativa de placa y el deterioro cognitivo [ 97 – 99 ]. La actividad de la cadena respiratoria se ve obstaculizada, específicamente en los complejos III y IV, lo que lleva a una disminución del metabolismo energético en las regiones afectadas [ 98 , 100 , 101 ]. Se sabe que el Aβ soluble se colocaliza en las mitocondrias y es importado por el complejo de importación mitocondrial, TIM/TOM [ 102 , 103 ]. Una vez en las mitocondrias, el Aβ actúa sobre varios objetivos moleculares, incluida la alcohol deshidrogenasa de unión a Aβ (ABAD) y el complejo IV, lo que desencadena el daño de la cadena de transporte de electrones y la posterior producción de ROS que conduce a una falla del potencial de membrana mitocondrial [ 104 , 63 , 105 , 103 ].
Además de dañar los componentes celulares, la generación de ROS inducida por Aβ también sirve para inducir la fragmentación mitocondrial como resultado de la s-nitrosilación de Drp1 [ 106 ]. Esta alteración en la dinámica mitocondrial es concomitante con disminuciones en el transporte mitocondrial axonal [ 104 ] Aβ también conduce a mayores niveles de Ca 2+ mitocondrial que pueden inducir la apertura de mPTP, liberando citocromo c y factor inductor de apoptosis (AIF) que desencadenan la inducción de vías de muerte celular [ 97 ]. Estas características apuntan a la sorprendente importancia clínica de la falla mitocondrial en la EA, así como un potente objetivo de terapias, como MB.
Se sabe bien que el MB promueve la actividad del complejo IV y la actividad mitocondrial y este efecto se extiende al cerebro con EA [ 107 , 26 ]. Uno de los mecanismos principales del MB es la promoción de la actividad del complejo IV a través del ciclo de electrones, pero un factor contribuyente es la regulación positiva de la síntesis del hemo [ 63 , 107 ]. En el modelo de rata con estreptozotocina (STZ) de EA, los aumentos en la producción del complejo IV y ATP se reflejaron en la mejora de los déficits cognitivos inducidos por el daño hipocampal [ 57 ]. Se ha demostrado que el MB disminuye los marcadores de estrés oxidativo en varios modelos de EA a través de métodos de ciclo de electrones y mediante la inhibición de mecanismos e interacciones posteriores [ 57 , 108 , 109 ].
Uno de estos métodos es la inhibición de la unión de Aβ-ABAD, lo que previene la producción asociada de ROS, la falla de MMP y la posterior muerte celular [ 109-111 , 103 ]. Promover la función mitocondrial puede prevenir la liberación citosólica de factores pro -muerte, pero MB puede llevar esta medida protectora un paso más allá al desactivar las caspasas a través de la oxidación de la cisteína funcional. MB también se dirige a otras características distintivas del trastorno, NFT y placas Aβ. Considerando la forma de retroalimentación progresiva de casi todos los aspectos de los mecanismos de la EA, estos mecanismos más amplios mitigan un mayor deterioro mitocondrial.
Otra característica significativa de la EA es la presencia de NFT, agregados de proteína tau hiperfosforilada (p-tau). Tau es una proteína asociada a microtúbulos que es altamente prevalente en el SNC, estabilizando los microtúbulos neuronales. Tau es un objetivo de fosforilación para quinasas como la quinasa 3 β de la glucógeno-sintasa (GSK3β), la quinasa c-Jun (JNK) y la quinasa 5 dependiente de ciclina (cdk5) [ 112 – 114 ]. Si bien la fosforilación de tau es un proceso fisiológico, la fosforilación excesiva de tau es patológica y contribuye a la degradación neuronal en la EA.
Cuando se fosforila excesivamente, la tau se disocia de los microtúbulos, lo que provoca su desestabilización y posterior disolución. Mientras tanto, la propia p-tau se agrega en ovillos [ 115 ]. Al alterar las redes de microtúbulos, se compromete la función celular y el transporte axonal, como los déficits de transporte mitocondrial antes mencionados [ 116 ]. También se obstaculiza la eliminación a través del proteasoma. Aunque la p-tau está muy ubiquitinilada, sigue acumulándose en exceso de lo que el proteasoma puede eliminar [ 117 , 116 ]. Se debate acaloradamente si la tauopatía precede a la patología Aβ o es una consecuencia de ella, al igual que su contribución exacta a la patología. Independientemente de ello, está claro que la agregación de tau desempeña un papel vital en la EA, y abordar su disfunción es un tema de estudio importante.
El trabajo pionero en la aplicación de MB a AD fue realizado por Claude Wischik, en donde su laboratorio encontró que MB revierte la agregación de tau al bloquear la unión tau-tau, aunque en dosis más altas que los niveles clínicamente relevantes [ 66 ]. MB y sus derivados disminuyeron la acumulación de filamentos de tau en un ensayo in vitro, que más tarde se encontró que se debía a la inhibición de la formación de filamentos en los péptidos de primera y cuarta repetición en el dominio de unión de microtúbulos tau [ 68 , 118 ]. A diferencia de otros colorantes relacionados, MB no se une ni se asocia con protofibrillas tau [ 119 ]. Previene la fibrilación al oxidar los residuos de cisteína en tau, lo que hace que formen una forma de monómero más estable que es resistente a la agregación [ 69 ].
Además, también se observó que MB ayudó a promover la eliminación de filamentos de tau al inducir la autofagia [ 65 ]. Múltiples estudios han encontrado que MB ayudó a reducir la carga de tau en diferentes modelos de ratones transgénicos de tauopatía durante el tratamiento a corto y largo plazo y que esta eliminación se asoció con la mejora de los déficits cognitivos [ 58 , 62 ]. Si se administra de forma preventiva, MB puede prevenir el deterioro cognitivo y la acumulación de tau en ratones transgénicos que expresan tau humana proagregante [ 120 ].
Los estudios que aplicaron MB a diferentes modelos animales arrojaron resultados interesantes, aunque algo contradictorios. MB no logró reducir la carga de tau en un modelo de pez cebra, aunque mostró resultados prometedores en la disminución de la agregación de huntingtina [ 121 ]. En un modelo de tauopatía transgénica de C. elegans, MB eliminó tau y promovió un transporte mitocondrial adecuado que estuvo acompañado de una mayor motilidad [ 122 ].
El MB se ha aplicado en ensayos clínicos en humanos, comenzando con un ensayo clínico de fase 2 para el MB bajo el nombre de “Rember”. El ensayo mostró mejoras cognitivas y del flujo sanguíneo cerebral en pacientes con EA leve a moderada [ 59 ]. Se desarrolló una variante de la molécula, leuco-metiltioninio bis (hidrometanosulfonato), o LMTM, que era más estable en la forma reducida [ 15 ]. Desafortunadamente, un estudio posterior de fase 3 de 15 meses de LMTM arrojó resultados negativos [ 14 ]. Próximamente se realizará otro estudio de LMTM en casos leves de EA. Estos estudios y otros pueden dilucidar aún más los mecanismos de protección del MB contra la EA y algún día podrían conducir a una traducción clínica.
3.3 Azul de metileno y enfermedad de Parkinson
La enfermedad de Parkinson (EP) es un trastorno neurodegenerativo progresivo que resulta de la muerte de neuronas dopaminérgicas en la sustancia negra (SN), que se presenta clínicamente como temblor, rigidez, alteración del movimiento, afecto facial plano y depresión [ 4 ]. La aparición de la enfermedad generalmente se presenta después de los 50 años, afectando a 100-300 de cada 100.000 personas, manifestando síntomas motores en la mayoría de los pacientes [ 123 ]. La EP disminuye la esperanza de vida de los pacientes, y la edad de aparición afecta significativamente el pronóstico; aquellos con inicio temprano tienen una esperanza de vida reducida de 10 años, mientras que aquellos con inicio tardío pierden alrededor de 5 años de esperanza de vida [ 124 ]. La mortalidad en la EP es comúnmente el resultado de caídas o infección respiratoria y edema, a menudo como resultado de la aspiración accidental de alimentos debido a la dificultad para tragar [ 125 – 127 ].
El tratamiento del Parkinson suele consistir en la administración de levodopa, un precursor de la dopamina que disminuye la rigidez y el temblor. Lamentablemente, la terapia con levodopa conlleva efectos secundarios neurológicos y conductuales, y normalmente se requieren dosis cada vez mayores y medicamentos secundarios a lo largo del tratamiento [ 128 ]. La levodopa, por vital que sea para los pacientes, es un tratamiento paliativo y no retrasa la progresión del trastorno. Por ello, es fundamental desarrollar terapias que puedan dirigirse a los mecanismos clave del daño neuronal para detener o revertir el curso de la enfermedad.
El sello histológico de la enfermedad de Parkinson es la presencia de cuerpos de Lewy y neuritas de Lewy, agregados intracelulares de la proteína α-sinucleína (α-syn) [ 4 ]. Estos agregados están fuertemente asociados con alteraciones en la expresión de la ligasa de ubiquitina E3, Parkin, que causa déficits en el control de calidad mitocondrial al impedir el tráfico de microtúbulos [ 129 ]. A-syn, cuando se transloca a las mitocondrias, puede perjudicar la función tanto del complejo I como del complejo IV de la cadena de transporte de electrones, lo que lleva a una disfunción mitocondrial progresiva que es potenciada por mitofagia deficiente [ 130 , 131 ]. Esta disfunción de la cadena de transporte de electrones es la base sobre la cual se generan dos de los modelos más utilizados de síntomas de EP: MPTP y rotenona, los cuales inhiben el complejo I [ 5 , 132 ]. Estas toxinas causan daños característicos a las neuronas dopaminérgicas del sistema nervioso central y manifiestan temblor parkinsoniano [ 132 ]. Como sucede con todas las enfermedades neurodegenerativas con un mecanismo mitocondrial, las estrategias terapéuticas dirigidas a dicha disfunción ofrecen una atractiva línea de investigación.
El trabajo seminal que determinó la capacidad de transferencia alternativa de electrones de MB se descubrió utilizando un modelo de rotenona de EP. Junto con la preservación antes mencionada de la función de la cadena de transporte de electrones, el tratamiento con MB se acompañó de mejoras conductuales en el temblor, la locomoción, la postura y las habilidades motoras [ 5 ]. Más allá de este estudio, la evidencia de la eficacia en la EP es indirecta, pero aún prometedora. Tanto la mitofagia como la autofagia general son inducidas por la administración de MB, lo que podría mitigar los déficits de control de calidad mitocondrial que conducen a la progresión del trastorno [ 51 , 65 ]. Además, las funciones antioxidantes y promotoras de la cadena de transporte de electrones establecidas de MB en las mitocondrias se dirigen a los complejos más atacados por la EP [ 27 , 5 , 133 ].
Con su bajo perfil de efectos secundarios, existe la posibilidad de que el MB administrado profilácticamente o en las primeras etapas de la EP pueda prevenir la progresión del trastorno debido a los efectos beneficiosos de los bajos niveles de H 2 O 2 generados por el MB en condiciones fisiológicas, mediados por la vía Nrf2/ARE, que está tentativamente implicada en la EP [ 134 , 135 , 30 ]. Finalmente, las propiedades IMAO del MB pueden proporcionar beneficios adicionales a un régimen continuo de levodopa, aunque el perfil de seguridad de esta combinación necesita ser investigado cuidadosamente [ 136 , 128 ]. La estrecha asociación del perfil de los efectos mitocondriales del MB y la mitopatía presente en la EP parece prometedora, destacando la necesidad de aplicar el MB a los diversos modelos animales químicos y genéticos de la EP.
El azul de metileno en la mejora cognitiva, el deterioro cognitivo relacionado con la edad y la neurogénesis
La mejora cognitiva a través de fármacos es una idea que ha captado la atención y la imaginación del público desde hace mucho tiempo. El concepto prevalece en la conciencia pública, y comunidades enteras en línea se centran en el concepto y la exploración de posibles potenciadores cognitivos, o nootrópicos. Los potenciadores cognitivos propuestos van desde suplementos de aminoácidos hasta suplementos herbales de uso clásico [ 151 , 152 ]. Los estimulantes prescritos clínicamente se suelen abusar, en particular en los campus universitarios, para este propósito, lo que aumenta el estigma público de los pacientes legítimos y la validez de los diagnósticos de trastorno por déficit de atención con hiperactividad en general [ 153 ]. Claramente, la mejora de la cognición es de gran interés para el público, así como para los investigadores. Las mejoras en la cognición podrían resultar a favor del bien público en términos de productividad y calidad de vida, especialmente con respecto a las poblaciones de edad avanzada. A la luz de esto, los compuestos disponibles a bajo costo que podrían apoyar o mejorar la cognición de manera segura son candidatos ideales. El MB, en este sentido, es por lo tanto un contendiente prometedor.
El concepto de mejora cognitiva a través de la modulación mitocondrial ha sido investigado cada vez más en los últimos años. El concepto general es que al mejorar la función mitocondrial y las defensas oxidativas, las neuronas pueden funcionar con mayor eficiencia y mantener una salud adecuada, mejorando la función basal y frenando el deterioro cognitivo asociado con la edad y la neurodegeneración [ 154 ]. El trabajo inicial de González-Lima ha demostrado que MB mejoró la retención de la memoria espacial junto con la función respiratoria mitocondrial de larga duración, mediada a través del complejo IV [ 26 ]. La regulación positiva a largo plazo de CCO puede estar relacionada con el aumento de la producción de H 2 O 2 sin formación de superóxido, a través de MB en condiciones fisiológicas, lo que lleva a la regulación positiva de Nrf2 / ARE [ 135 ]. En un estudio humano, la administración de MB aumentó la reactividad cerebrovascular en la tarea de vigilancia psicomotora y una prueba de memoria a corto plazo. Esto fue acompañado con modestas mejoras en el rendimiento en la prueba de memoria a corto plazo [ 155 ]. Estos beneficios correlacionados con la función mitocondrial están corroborados por experimentos que muestran una cognición mejorada de manera similar con la fotobiomodulación, la estimulación del complejo IV con irradiación láser transcraneal cercana al infrarrojo [ 156 ].
El trastorno metabólico es una observación bien establecida en los cerebros de los ancianos, especialmente en regiones clásicamente asociadas con funciones cerebrales superiores [ 157–159 ]. El daño mitocondrial se acumula con el tiempo y contribuye progresivamente al deterioro neuronal a medida que uno envejece, de manera muy similar a las condiciones neurodegenerativas [ 158 ]. Estas agresiones mitocondriales incluyen la ruptura de la cadena de transferencia de electrones, mitofagia deficiente y un cambio hacia la fisión excesiva, mutaciones del ADNmt y producción excesiva de ROS [ 158 ]. Estas características están asociadas con déficits cognitivos, en los ancianos, al igual que en aquellos con EA, GCI, trastorno depresivo mayor y otras afecciones. Como tal, proteger la salud mitocondrial puede proporcionar cierta medida de protección contra la demencia u otras formas de déficits cognitivos relacionados con la edad. MB, como se detalló anteriormente, puede obstaculizar o prevenir muchas de estas agresiones mitocondriales y puede tener potencial, de esta manera, para proteger la cognición contra los estragos metabólicos de la edad avanzada. Esta posibilidad, hasta el momento, no ha sido investigada formalmente y sigue siendo una cuestión abierta y un campo de investigación científica.
Se acepta ampliamente que la neurogénesis ocurre durante la edad adulta, tanto como un proceso fisiológico como una respuesta a una lesión [ 160 ]. Después del desarrollo, la neurogénesis se limita a tres regiones del cerebro, el bulbo olfatorio, la zona subventricular cortical (SVZ) y la zona subgranular (SGZ) del giro dentado (DG) del hipocampo. El deterioro de este proceso se asocia con la interrupción de la consolidación de la memoria y la regulación del estado de ánimo, y está implicado en la neurodegeneración y el deterioro cognitivo relacionado con la edad, ya que la neurogénesis y los procesos relacionados disminuyen drásticamente en la vejez [ 161 ]. Como tal, estimular y mantener la neurogénesis es un objetivo atractivo para la demencia relacionada con la edad, considerando las características compartidas sustanciales con otras formas patológicas de neurodegeneración.
La neurogénesis también se estimula e inhibe simultáneamente en las condiciones que se producen después de una lesión cerebral [ 162 ]. En modelos animales de accidente cerebrovascular, la neurogénesis se promueve tanto en la SVZ como en la SGZ junto con la liberación de eritropoyetina y factores de crecimiento epitelial vascular y epidérmico que inducen la proliferación de células progenitoras neuronales (NPC) [ 162 – 164 ]. Las neuronas recién formadas migran a la región lesionada e intentan incorporarse funcionalmente en el circuito neuronal existente para facilitar la reparación [ 165 ]. La reparación neuronal mediante la estimulación de la neurogénesis es atractiva, pero está limitada por la respuesta microambiental a la lesión. La neuroinflamación crónica y la activación glial, ambas características asociadas con el avance de la edad [ 166 ] son compartidas por la mayoría de las condiciones neurodegenerativas, y ambos factores atrofian la neurogénesis [ 166 ] [ 160 , 167 – 171 ]. Este déficit en la neurogénesis puede mitigarse con tratamientos antiinflamatorios [ 172 ]. Las ROS generadas por la glía reactiva también dañan y agotan los depósitos de células madre neuronales [ 173 ]. La adición de un antioxidante, al igual que el bloqueo antiinflamatorio, evitó el agotamiento de las neuronas recién formadas [ 174 ]. Además, la integridad de las mitocondrias es crucial para la neurogénesis y, por lo tanto, se regula positivamente durante la proliferación y la diferenciación [ 175 ] . Los déficits de mitocondrias, en gran medida realizados en experimentos cíbridos, muestran deterioros marcados tanto en la proliferación como en la diferenciación [ 176-178 ] .
Pocos estudios han aplicado MB a la neurogénesis, pero aquellos que lo han hecho, así como las acciones generales de MB, sugieren un potencial de efectos beneficiosos. La investigación de GCI mencionada anteriormente que aplicó MB a un modelo de lechones reveló que MB sobrerregulaba genes relacionados con la neurogénesis, incluyendo sinaptogénesis, crecimiento de neuritas y factores pro-supervivencia [ 91 ]. MB sobrerregulaba negativamente la proliferación de NPC de rata al regular negativamente la expresión de ciclina y la actividad de mTOR. Esto parece contradictorio, pero la proliferación excesiva agota el acervo de NPC, disminuyendo la capacidad futura para la neurogénesis. MB tampoco afectó la diferenciación neuronal. Estos resultados, sin embargo, se observaron en ratas del día 1 postnatal, por lo que no está claro si este mecanismo protector es relevante en el cerebro adulto lesionado [ 179 ]. El trabajo en nuestro laboratorio encontró que MB administrado después de un accidente cerebrovascular fototrombótico promovió la neurogénesis. Además, sobrerregulaba la inflamación, la gliosis y mejoraba la función mitocondrial, todas características que contribuyen a los déficits neurogénicos. Esto nos lleva a creer que el MB puede posiblemente promover o apoyar la neurogénesis después de una lesión al hacer que el microambiente local sea más propicio para las neuronas recién formadas mientras apoya su eficiencia mitocondrial [ 54 ]. Si bien sostener la supervivencia de las neuronas recién formadas es primordial, apoyar su migración es igualmente imperativo para sostener la reparación neuronal. Una vez más, la evidencia es limitada, pero la evidencia reciente indica que el MB podría modular la migración de células madre neuronales adultas murinas en un ensayo de migración celular [ 180 ]. Se necesita mucha más evidencia para sacar una conclusión sólida sobre el efecto y el papel del MB en la promoción de la neurogénesis adulta y la salud mitocondrial en la vejez. Si se valida, el MB puede servir como una herramienta multifacética para preservar la cognición y la calidad de vida de la población de edad avanzada, un grupo demográfico vital en una sociedad que envejece.

Descargar libros desde https://cienciaysaludnatural.com/recursos
Efectos adversos y contraindicaciones del azul de metileno
El MB es claramente prometedor como tratamiento terapéutico para muchas enfermedades, pero, como cualquier fármaco, conlleva una serie de limitaciones. Las propiedades vasomoduladoras del MB son útiles en dosis terapéuticas, pero conllevan riesgos en concentraciones más altas. Estos riesgos van desde el aumento de la presión arterial y la resistencia vascular hasta alteraciones de la función cardíaca y la ritmicidad [ 2 ]. Asimismo, el MB puede influir en las mediciones de oximetría de pulso, lo que dificulta un diagnóstico adecuado.
Otro efecto tóxico es la inducción de anemia en individuos con deficiencia de glucosa-6-fosfato deshidrogenasa. Los neonatos también son susceptibles a la anemia y muchos otros efectos adversos como respuesta al MB [ 2 ]. En el neonato, el MB puede inducir hiperbilirrubinemia, que se presenta como ictericia [ 181 ]. Esto es bastante desafortunado, ya que el tratamiento aceptado para la hiperbilirrubinemia es la fototerapia, que activa la actividad fotodinámica del MB. Esto genera especies de oxígeno excitadas, que causan un daño significativo a la epidermis [ 182 ]. Por lo tanto, en cualquier aplicación, el MB debe tratarse con el cuidado de cualquier otro fotosensibilizador. En el neonato, el tratamiento con MB también puede conducir, paradójicamente, a metahemoglobinemia [ 183 ].
El efecto adverso final, y el más relevante clínicamente en el paciente adulto, está relacionado con el uso de medicamentos psiquiátricos comunes, los inhibidores selectivos de la recaptación de serotonina (ISRS). El MB es un IMAO potente que, junto con un ISRS, puede potenciar el síndrome serotoninérgico, una emergencia médica potencialmente mortal [ 184–186 ] . Considerando la ubicuidad de los pacientes que toman ISRS, es prudente realizar pruebas de detección de estos medicamentos antes de administrar MB. De todos modos, el hecho de que los riesgos del MB sean relativamente bajos y razonablemente predecibles es un buen augurio para la adaptación en la clínica.
7. Conclusiones
Los trastornos neurodegenerativos son un desafío enorme en la medicina, que exige un alto precio, tanto humano como financiero. La relativa escasez de opciones terapéuticas es intrínsecamente desalentadora, pero es esta misma vacante la que debería estimular la innovación. La lucha de estos pacientes motiva a los investigadores dedicados, y la naturaleza inexplorada de este mercado debería alentar a los inversores a apoyar estos esfuerzos. Aunque estas ambiciones pueden verse empañadas por un historial menos que estelar, hay esperanza en empresas de menor riesgo en forma de productos farmacéuticos reutilizados, como el MB. El excelente historial de seguridad del MB está bien establecido por un siglo de uso médico. Su papel recientemente explorado como potenciador mitocondrial y antioxidante de reciclaje es clave para su potencial como agente terapéutico. Con resultados alentadores en ensayos preclínicos de algunas de las enfermedades neurodegenerativas más discapacitantes, el MB debería investigarse con renovado entusiasmo en los próximos años. Si estos resultados se validan en futuros estudios en humanos, el MB podría demostrar ser un agente versátil que podría mejorar la salud de los pacientes que sufren la carga de trastornos neurodegenerativos y lesiones cerebrales, brindándoles a ellos y a sus seres queridos alivio y una mejor calidad de vida.
Ver Parte I: Azul de Metileno, guia básica
Colabore por favor con nosotros para que podamos incluir mas información y llegar a más personas: contribución en mercado pago o paypal por única vez, Muchas Gracias!
Via PAYPAL: Euros o dólares click aqui
ARGENTINA 10.000$ar https://mpago.la/1srgnEY
5.000$ar https://mpago.la/1qzSyt9
1.000$ar https://mpago.la/1Q1NEKM
Solicite nuestro CBU contactenos
Referencias
- 1.Schirmer RH, Adler H, Pickhardt M, Mandelkow E. Lest we forget you–methylene blue…. Neurobiology of aging. 2011;32(12):2325 e2327–2316. doi: 10.1016/j.neurobiolaging.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Ginimuge PR, Jyothi SD. Methylene blue: revisited. Journal of anaesthesiology, clinical pharmacology. 2010;26(4):517–520. [PMC free article] [PubMed] [Google Scholar]
- 3.Stawicki SP, Sims C, Sarani B, Grossman MD, Gracias VH. Methylene blue and vasoplegia: who, when, and how? Mini reviews in medicinal chemistry. 2008;8(5):472–490. doi: 10.2174/138955708784223477. [DOI] [PubMed] [Google Scholar]
- 4.Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L, Song BJ. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain research. 2016;1637:34–55. doi: 10.1016/j.brainres.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang SH, Li W, Sumien N, Forster M, Simpkins JW, Liu R. Alternative mitochondrial electron transfer for the treatment of neurodegenerative diseases and cancers: Methylene blue connects the dots. Prog Neurobiol. 2015 doi: 10.1016/j.pneurobio.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guttman PE. On the Action of Methylene Blue on Malaria. In: Himmelwelt F, editor. The Collected Papers of Paul Ehrlich: Chemotherapy, vol III. Elsivier; 1891. pp. 15–20. [Google Scholar]
- 7.Schaefer B, E P. Natural Products in the Chemical Industry. Springer; 2015. Illustrated edn. [Google Scholar]
- 8.Hughes DE, L E. The Use of Methylene Blue as a Seditive. In: Curtin Roland G, D EH., editors. Philadelphia Hospital Reports. Vol. 4. Detre & Blackburn: 1901. pp. 272–282. [Google Scholar]
- 9.Gillman PK. CNS toxicity involving methylene blue: the exemplar for understanding and predicting drug interactions that precipitate serotonin toxicity. J Psychopharmacol. 2011;25(3):429–436. doi: 10.1177/0269881109359098. [DOI] [PubMed] [Google Scholar]
- 10.Eroglu L, Caglayan B. Anxiolytic and antidepressant properties of methylene blue in animal models. Pharmacological research. 1997;36(5):381–385. doi: 10.1006/phrs.1997.0245. [DOI] [PubMed] [Google Scholar]
- 11.Alda M, McKinnon M, Blagdon R, Garnham J, MacLellan S, O’Donovan C, Hajek T, Nair C, Dursun S, MacQueen G. Methylene blue treatment for residual symptoms of bipolar disorder: randomised crossover study. The British journal of psychiatry: the journal of mental science. 2017;210(1):54–60. doi: 10.1192/bjp.bp.115.173930. [DOI] [PubMed] [Google Scholar]
- 12.Peter C, Hongwan D, Kupfer A, Lauterburg BH. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur J Clin Pharmacol. 2000;56(3):247–250. doi: 10.1007/s002280000124. [DOI] [PubMed] [Google Scholar]
- 13.DiSanto AR, Wagner JG. Pharmacokinetics of highly ionized drugs. 3. Methylene blue–blood levels in the dog and tissue levels in the rat following intravenous administration. J Pharm Sci. 1972;61(7):1090–1094. doi: 10.1002/jps.2600610711. [DOI] [PubMed] [Google Scholar]
- 14.Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, Schelter BO, Davis CS, Staff RT, Bracoud L, Shamsi K, Storey JM, Harrington CR, Wischik CM. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388(10062):2873–2884. doi: 10.1016/S0140-6736(16)31275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baddeley TC, McCaffrey J, Storey JM, Cheung JK, Melis V, Horsley D, Harrington CR, Wischik CM. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer’s disease. J Pharmacol Exp Ther. 2015;352(1):110–118. doi: 10.1124/jpet.114.219352. [DOI] [PubMed] [Google Scholar]
- 16.Coleman MD, Coleman NA. Drug-induced methaemoglobinaemia. Treatment issues. Drug safety. 1996;14(6):394–405. doi: 10.2165/00002018-199614060-00005. [DOI] [PubMed] [Google Scholar]
- 17.Bradberry SM. Occupational methaemoglobinaemia. Mechanisms of production, features, diagnosis and management including the use of methylene blue. Toxicological reviews. 2003;22(1):13–27. doi: 10.2165/00139709-200322010-00003. [DOI] [PubMed] [Google Scholar]
- 18.Cawein M, Behlen CH, 2nd, Lappat EJ, Cohn JE. Hereditary Diaphorase Deficiency and Methemoglobinemia. Archives of internal medicine. 1964;113:578–585. doi: 10.1001/archinte.1964.00280100086014. [DOI] [PubMed] [Google Scholar]
- 19.Oz M, Lorke DE, Hasan M, Petroianu GA. Cellular and molecular actions of Methylene Blue in the nervous system. Medicinal research reviews. 2011;31(1):93–117. doi: 10.1002/med.20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alici-Evcimen Y, Breitbart WS. Ifosfamide neuropsychiatric toxicity in patients with cancer. Psycho-oncology. 2007;16(10):956–960. doi: 10.1002/pon.1161. [DOI] [PubMed] [Google Scholar]
- 21.Shanmugam G. Vasoplegic syndrome–the role of methylene blue. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2005;28(5):705–710. doi: 10.1016/j.ejcts.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 22.2015 WHO Model List of Essential Medicines. 2015 http://www.who.int/medicines/publications/essentialmedicines/en/
- 23.Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. The Journal of biological chemistry. 2011;286(18):16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opii WO, Nukala VN, Sultana R, Pandya JD, Day KM, Merchant ML, Klein JB, Sullivan PG, Butterfield DA. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. Journal of neurotrauma. 2007;24(5):772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- 25.Wong-Riley MT. Bigenomic regulation of cytochrome c oxidase in neurons and the tight coupling between neuronal activity and energy metabolism. Advances in experimental medicine and biology. 2012;748:283–304. doi: 10.1007/978-1-4614-3573-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F. Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacology, biochemistry, and behavior. 2004;77(1):175–181. doi: 10.1016/j.pbb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Gureev AP, Syromyatnikov MY, Gorbacheva TM, Starkov AA, Popov VN. Methylene blue improves sensorimotor phenotype and decreases anxiety in parallel with activating brain mitochondria biogenesis in mid-age mice. Neurosci Res. 2016;113:19–27. doi: 10.1016/j.neures.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Wu HM, Lee CG, Hwang SJ, Kim SG. Mitigation of carbon tetrachloride-induced hepatic injury by methylene blue, a repurposed drug, is mediated by dual inhibition of GSK3beta downstream of PKA. Br J Pharmacol. 2014;171(11):2790–2802. doi: 10.1111/bph.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre-Ghiso J, Flores SC, Melendez JA. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280(17):16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- 30.Stack C, Jainuddin S, Elipenahli C, Gerges M, Starkova N, Starkov AA, Jove M, Portero-Otin M, Launay N, Pujol A, Kaidery NA, Thomas B, Tampellini D, Beal MF, Dumont M. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum Mol Genet. 2014;23(14):3716–3732. doi: 10.1093/hmg/ddu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends in neurosciences. 2013;36(10):587–597. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32(7):1222–1232. doi: 10.1038/jcbfm.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du F, Zhu XH, Zhang Y, Friedman M, Zhang N, Ugurbil K, Chen W. Tightly coupled brain activity and cerebral ATP metabolic rate. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(17):6409–6414. doi: 10.1073/pnas.0710766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth AD, Nunez MT. Oligodendrocytes: Functioning in a Delicate Balance Between High Metabolic Requirements and Oxidative Damage. Advances in experimental medicine and biology. 2016;949:167–181. doi: 10.1007/978-3-319-40764-7_8. [DOI] [PubMed] [Google Scholar]
- 36.Song BJ, Akbar M, Abdelmegeed MA, Byun K, Lee B, Yoon SK, Hardwick JP. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox biology. 2014;3:109–123. doi: 10.1016/j.redox.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. Journal of the neurological sciences. 2005;233(1–2):145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 38.Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. The Journal of pharmacology and experimental therapeutics. 2012;342(3):619–630. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanderson TH, Raghunayakula S, Kumar R. Release of mitochondrial Opa1 following oxidative stress in HT22 cells. Molecular and cellular neurosciences. 2015;64:116–122. doi: 10.1016/j.mcn.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain research reviews. 2011;67(1–2):103–118. doi: 10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 42.Giannoccaro MP, La Morgia C, Rizzo G, Carelli V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society. 2017;32(3):346–363. doi: 10.1002/mds.26966. [DOI] [PubMed] [Google Scholar]
- 43.Lu Q, Tucker D, Dong Y, Zhao N, Zhang Q. Neuroprotective and Functional Improvement Effects of Methylene Blue in Global Cerebral Ischemia. Mol Neurobiol. 2016;53(8):5344–5355. doi: 10.1007/s12035-015-9455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryou MG, Choudhury GR, Li W, Winters A, Yuan F, Liu R, Yang SH. Methylene blue-induced neuronal protective mechanism against hypoxia-reoxygenation stress. Neuroscience. 2015;301:193–203. doi: 10.1016/j.neuroscience.2015.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiklund L, Basu S, Miclescu A, Wiklund P, Ronquist G, Sharma HS. Neuro- and cardioprotective effects of blockade of nitric oxide action by administration of methylene blue. Ann N Y Acad Sci. 2007;1122:231–244. doi: 10.1196/annals.1403.016. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Yang R, Li P, Lu H, Hao J, Tucker D, Zhang Q. Combination Treatment with Methylene Blue and Hypothermia in Global Cerebral Ischemia. Mol Neurobiol. 2017 doi: 10.1007/s12035-017-0470-1. [DOI] [PubMed] [Google Scholar]
- 47.Wiklund L, Zoerner F, Semenas E, Miclescu A, Basu S, Sharma HS. Improved neuroprotective effect of methylene blue with hypothermia after porcine cardiac arrest. Acta Anaesthesiol Scand. 2013;57(8):1073–1082. doi: 10.1111/aas.12106. [DOI] [PubMed] [Google Scholar]
- 48.Pakavathkumar P, Sharma G, Kaushal V, Foveau B, LeBlanc AC. Methylene Blue Inhibits Caspases by Oxidation of the Catalytic Cysteine. Sci Rep. 2015;5:13730. doi: 10.1038/srep13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weingartner R, Oliveira E, Oliveira ES, Sant’Anna UL, Oliveira RP, Azambuja LA, Friedman G. Blockade of the action of nitric oxide in human septic shock increases systemic vascular resistance and has detrimental effects on pulmonary function after a short infusion of methylene blue. Braz J Med Biol Res. 1999;32(12):1505–1513. doi: 10.1590/s0100-879×1999001200009. [DOI] [PubMed] [Google Scholar]
- 50.Jiang Z, Watts LT, Huang S, Shen Q, Rodriguez P, Chen C, Zhou C, Duong TQ. The Effects of Methylene Blue on Autophagy and Apoptosis in MRI-Defined Normal Tissue, Ischemic Penumbra and Ischemic Core. PLoS One. 2015;10(6):e0131929. doi: 10.1371/journal.pone.0131929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Y, He YL, Zhao T, Huang X, Wu KW, Liu SH, Zhao YQ, Fan M, Wu LY, Zhu LL. Methylene Blue Reduces Acute Cerebral Ischemic Injury via the Induction of Mitophagy. Mol Med. 2015;21:420–429. doi: 10.2119/molmed.2015.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez P, Jiang Z, Huang S, Shen Q, Duong TQ. Methylene blue treatment delays progression of perfusion-diffusion mismatch to infarct in permanent ischemic stroke. Brain Res. 2014;1588:144–149. doi: 10.1016/j.brainres.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen Q, Du F, Huang S, Rodriguez P, Watts LT, Duong TQ. Neuroprotective efficacy of methylene blue in ischemic stroke: an MRI study. PLoS One. 2013;8(11):e79833. doi: 10.1371/journal.pone.0079833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed ME, Tucker D, Dong Y, Lu Y, Zhao N, Wang R, Zhang Q. Methylene Blue promotes cortical neurogenesis and ameliorates behavioral deficit after photothrombotic stroke in rats. Neuroscience. 2016;336:39–48. doi: 10.1016/j.neuroscience.2016.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu H, Li J, Wang Z, Feng M, Shen Y, Cao S, Li T, Peng Y, Fan L, Chen J, Gu C, Yan F, Wang L, Chen G. Methylene blue attenuates neuroinflammation after subarachnoid hemorrhage in rats through the Akt/GSK-3beta/MEF2D signaling pathway. Brain Behav Immun. 2017 doi: 10.1016/j.bbi.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez P, Zhao J, Milman B, Tiwari YV, Duong TQ. Methylene blue and normobaric hyperoxia combination therapy in experimental ischemic stroke. Brain Behav. 2016;6(7):e00478. doi: 10.1002/brb3.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li L, Qin L, Lu HL, Li PJ, Song YJ, Yang RL. Methylene blue improves streptozotocin-induced memory deficit by restoring mitochondrial function in rats. Brain Res. 2017;1657:208–214. doi: 10.1016/j.brainres.2016.12.024. [DOI] [PubMed] [Google Scholar]
- 58.Melis V, Magbagbeolu M, Rickard JE, Horsley D, Davidson K, Harrington KA, Goatman K, Goatman EA, Deiana S, Close SP, Zabke C, Stamer K, Dietze S, Schwab K, Storey JM, Harrington CR, Wischik CM, Theuring F, Riedel G. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol. 2015;26(4):353–368. doi: 10.1097/FBP.0000000000000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44(2):705–720. doi: 10.3233/JAD-142874. [DOI] [PubMed] [Google Scholar]
- 60.Mori T, Koyama N, Segawa T, Maeda M, Maruyama N, Kinoshita N, Hou H, Tan J, Town T. Methylene blue modulates beta-secretase, reverses cerebral amyloidosis, and improves cognition in transgenic mice. J Biol Chem. 2014;289(44):30303–30317. doi: 10.1074/jbc.M114.568212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paban V, Manrique C, Filali M, Maunoir-Regimbal S, Fauvelle F, Alescio-Lautier B. Therapeutic and preventive effects of methylene blue on Alzheimer’s disease pathology in a transgenic mouse model. Neuropharmacology. 2014;76(Pt A):68–79. doi: 10.1016/j.neuropharm.2013.06.033. [DOI] [PubMed] [Google Scholar]
- 62.Hosokawa M, Arai T, Masuda-Suzukake M, Nonaka T, Yamashita M, Akiyama H, Hasegawa M. Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS One. 2012;7(12):e52389. doi: 10.1371/journal.pone.0052389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis. 2010;20(Suppl 2):S439–452. doi: 10.3233/JAD-2010-100414. [DOI] [PubMed] [Google Scholar]
- 64.Medina DX, Caccamo A, Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21(2):140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff KE. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8(4):609–622. doi: 10.4161/auto.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93(20):11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, Glabe CG. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46(30):8850–8860. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- 68.Hattori M, Sugino E, Minoura K, In Y, Sumida M, Taniguchi T, Tomoo K, Ishida T. Different inhibitory response of cyanidin and methylene blue for filament formation of tau microtubule-binding domain. Biochemical and biophysical research communications. 2008;374(1):158–163. doi: 10.1016/j.bbrc.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 69.Crowe A, James MJ, Lee VM, Smith AB, 3rd, Trojanowski JQ, Ballatore C, Brunden KR. Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation. J Biol Chem. 2013;288(16):11024–11037. doi: 10.1074/jbc.M112.436006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao M, Liang F, Xu H, Yan W, Zhang J. Methylene blue exerts a neuroprotective effect against traumatic brain injury by promoting autophagy and inhibiting microglial activation. Mol Med Rep. 2016;13(1):13–20. doi: 10.3892/mmr.2015.4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Talley Watts L, Long JA, Boggs RC, Manga H, Huang S, Shen Q, Duong TQ. Delayed Methylene Blue Improves Lesion Volume, Multi-Parametric Quantitative Magnetic Resonance Imaging Measurements, and Behavioral Outcome after Traumatic Brain Injury. J Neurotrauma. 2016;33(2):194–202. doi: 10.1089/neu.2015.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Talley Watts L, Long JA, Chemello J, Van Koughnet S, Fernandez A, Huang S, Shen Q, Duong TQ. Methylene blue is neuroprotective against mild traumatic brain injury. J Neurotrauma. 2014;31(11):1063–1071. doi: 10.1089/neu.2013.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fenn AM, Skendelas JP, Moussa DN, Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP. Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J Neurotrauma. 2015;32(2):127–138. doi: 10.1089/neu.2014.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mangus DB, Huang L, Applegate PM, Gatling JW, Zhang J, Applegate RL., 2nd A systematic review of neuroprotective strategies after cardiac arrest: from bench to bedside (Part I – Protection via specific pathways) Medical gas research. 2014;4:9. doi: 10.1186/2045-9912-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schneider A, Bottiger BW, Popp E. Cerebral resuscitation after cardiocirculatory arrest. Anesthesia and analgesia. 2009;108(3):971–979. doi: 10.1213/ane.0b013e318193ca99. [DOI] [PubMed] [Google Scholar]
- 76.Kim YM, Yim HW, Jeong SH, Klem ML, Callaway CW. Does therapeutic hypothermia benefit adult cardiac arrest patients presenting with non-shockable initial rhythms?: A systematic review and meta-analysis of randomized and non-randomized studies. Resuscitation. 2012;83(2):188–196. doi: 10.1016/j.resuscitation.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 77.Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Bbttiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, Kern KB, Laurent I, Longstreth WT, Merchant RM, Morley P, Morrison LJ, Nadkarni V, Peberdy MA, Rivers EP, Rodriguez-Nunez A, Sellke FW, Spaulding C, Sunde K, Vanden Hoek T. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication: a scientific statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke (Part II) International emergency nursing. 2010;18(1):8–28. doi: 10.1016/j.ienj.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 78.Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloeck WG, Billi J, Bottiger BW, Okada K, Reyes C, Shuster M, Steen PA, Weil MH, Wenzel V, Carli P, Atkins D. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108(1):118–121. doi: 10.1161/01.CIR.0000079019.02601.90. [DOI] [PubMed] [Google Scholar]
- 79.Scirica BM. Therapeutic hypothermia after cardiac arrest. Circulation. 2013;127(2):244–250. doi: 10.1161/CIRCULATIONAHA.111.076851. [DOI] [PubMed] [Google Scholar]
- 80.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 81.Victor Li, X B, Paul Szelemej, Jiming Kong. Delayed Neuronal Death in Ischemic Stroke: Molecular Pathways. In: Balestrino DM, editor. Advances in the Preclinical Study of Ischemic Stroke. InTech. [Google Scholar]
- 82.Kim J, Park JE, Nahrendorf M, Kim DE. Direct Thrombus Imaging in Stroke. Journal of stroke. 2016;18(3):286–296. doi: 10.5853/jos.2016.00906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kirmani JF, Alkawi A, Panezai S, Gizzi M. Advances in thrombolytics for treatment of acute ischemic stroke. Neurology. 2012;79(13 Suppl 1):S119–125. doi: 10.1212/WNL.0b013e3182695882. [DOI] [PubMed] [Google Scholar]
- 84.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. The New England journal of medicine. 1995;333(24):1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 85.Ferrer I. Apoptosis: future targets for neuroprotective strategies. Cerebrovasc Dis. 2006;21(Suppl 2):9–20. doi: 10.1159/000091699. [DOI] [PubMed] [Google Scholar]
- 86.Siesjo BK. Cell damage in the brain: a speculative synthesis. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1981;1(2):155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- 87.Kaur C, Ling EA. Blood brain barrier in hypoxic-ischemic conditions. Current neurovascular research. 2008;5(1):71–81. doi: 10.2174/156720208783565645. [DOI] [PubMed] [Google Scholar]
- 88.Giulian D, Li J, Leara B, Keenen C. Phagocytic microglia release cytokines and cytotoxins that regulate the survival of astrocytes and neurons in culture. Neurochemistry international. 1994;25(3):227–233. doi: 10.1016/0197-0186(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 89.Bothe HW, Bosma HJ, Hofer H, Hossmann KA, Angermeier WF. Selective vulnerability of hippocampus and disturbances of memory storage after mild unilateral ischemia of gerbil brain. Stroke. 1986;17(6):1160–1163. doi: 10.1161/01.str.17.6.1160. [DOI] [PubMed] [Google Scholar]
- 90.Sharma HS, Miclescu A, Wiklund L. Cardiac arrest-induced regional blood-brain barrier breakdown, edema formation and brain pathology: a light and electron microscopic study on a new model for neurodegeneration and neuroprotection in porcine brain. J Neural Transm (Vienna) 2011;118(1):87–114. doi: 10.1007/s00702-010-0486-4. [DOI] [PubMed] [Google Scholar]
- 91.Martijn C, Wiklund L. Effect of methylene blue on the genomic response to reperfusion injury induced by cardiac arrest and cardiopulmonary resuscitation in porcine brain. BMC medical genomics. 2010;3:27. doi: 10.1186/1755-8794-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xie L, Li W, Winters A, Yuan F, Jin K, Yang S. Methylene blue induces macroautophagy through 5′ adenosine monophosphate-activated protein kinase pathway to protect neurons from serum deprivation. Frontiers in cellular neuroscience. 2013;7:56. doi: 10.3389/fncel.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fang Q, Yan X, Li S, Sun Y, Xu L, Shi Z, Wu M, Lu Y, Dong L, Liu R, Yuan F, Yang SH. Methylene Blue Ameliorates Ischemia/Reperfusion-Induced Cerebral Edema: An MRI and Transmission Electron Microscope Study. Acta neurochirurgica Supplement. 2016;121:227–236. doi: 10.1007/978-3-319-18497-5_41. [DOI] [PubMed] [Google Scholar]
- 94.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. Journal of cell science. 2007;120(Pt 23):4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 95.Arrazola MS, Silva-Alvarez C, Inestrosa NC. How the Wnt signaling pathway protects from neurodegeneration: the mitochondrial scenario. Frontiers in cellular neuroscience. 2015;9:166. doi: 10.3389/fncel.2015.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Irwin JA, Wong HE, Kwon I. Different fates of Alzheimer’s disease amyloid-beta fibrils remodeled by biocompatible small molecules. Biomacromolecules. 2013;14(1):264–274. doi: 10.1021/bm3016994. [DOI] [PubMed] [Google Scholar]
- 97.Supnet C, Bezprozvanny I. Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2010;20(Suppl 2):S487–498. doi: 10.3233/JAD-2010-100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(34):14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21(9):3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Human molecular genetics. 2006;15(9):1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 101.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. European journal of nuclear medicine and molecular imaging. 2005;32(4):486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- 102.Devi L, Anandatheerthavarada HK. Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochimica et biophysica acta. 2010;1802(1):11–19. doi: 10.1016/j.bbadis.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 104.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Human molecular genetics. 2011;20(23):4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pratico D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends in pharmacological sciences. 2008;29(12):609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 106.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Atamna H. Amino acids variations in amyloid-beta peptides, mitochondrial dysfunction, and new therapies for Alzheimer’s disease. Journal of bioenergetics and biomembranes. 2009;41(5):457–464. doi: 10.1007/s10863-009-9246-2. [DOI] [PubMed] [Google Scholar]
- 108.Violet M, Chauderlier A, Delattre L, Tardivel M, Chouala MS, Sultan A, Marciniak E, Humez S, Binder L, Kayed R, Lefebvre B, Bonnefoy E, Buee L, Galas MC. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiology of disease. 2015;82:540–551. doi: 10.1016/j.nbd.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 109.Zakaria A, Hamdi N, Abdel-Kader RM. Methylene Blue Improves Brain Mitochondrial ABAD Functions and Decreases Abeta in a Neuroinflammatory Alzheimer’s Disease Mouse Model. Molecular neurobiology. 2016;53(2):1220–1228. doi: 10.1007/s12035-014-9088-8. [DOI] [PubMed] [Google Scholar]
- 110.Lim YA, Grimm A, Giese M, Mensah-Nyagan AG, Villafranca JE, Ittner LM, Eckert A, Gotz J. Inhibition of the mitochondrial enzyme ABAD restores the amyloid-beta-mediated deregulation of estradiol. PloS one. 2011;6(12):e28887. doi: 10.1371/journal.pone.0028887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, Trillat AC, Stern DM, Arancio O, Yan SS. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19(6):597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 112.Yarza R, Vela S, Solas M, Ramirez MJ. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Frontiers in pharmacology. 2015;6:321. doi: 10.3389/fphar.2015.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B, Laferla F, Gallego-Gomez JC, Kosik KS, Cardona-Gomez GP. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(42):13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. Journal of neurochemistry. 2008;104(6):1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G. Tau protein modifications and interactions: their role in function and dysfunction. International journal of molecular sciences. 2014;15(3):4671–4713. doi: 10.3390/ijms15034671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rodriguez-Martin T, Cuchillo-Ibanez I, Noble W, Nyenya F, Anderton BH, Hanger DP. Tau phosphorylation affects its axonal transport and degradation. Neurobiology of aging. 2013;34(9):2146–2157. doi: 10.1016/j.neurobiolaging.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sulistio YA, Heese K. The Ubiquitin-Proteasome System and Molecular Chaperone Deregulation in Alzheimer’s Disease. Molecular neurobiology. 2016;53(2):905–931. doi: 10.1007/s12035-014-9063-4. [DOI] [PubMed] [Google Scholar]
- 118.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. The Journal of biological chemistry. 2005;280(9):7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 119.Lira-De Leon KI, Garcia-Gutierrez P, Serratos IN, Palomera-Cardenas M, Figueroa-Corona Mdel P, Campos-Pena V, Meraz-Rios MA. Molecular mechanism of tau aggregation induced by anionic and cationic dyes. Journal of Alzheimer’s disease: JAD. 2013;35(2):319–334. doi: 10.3233/JAD-121765. [DOI] [PubMed] [Google Scholar]
- 120.Hochgrafe K, Sydow A, Matenia D, Cadinu D, Konen S, Petrova O, Pickhardt M, Goll P, Morellini F, Mandelkow E, Mandelkow EM. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta neuropathologica communications. 2015;3:25. doi: 10.1186/s40478-015-0204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van Bebber F, Paquet D, Hruscha A, Schmid B, Haass C. Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiology of disease. 2010;39(3):265–271. doi: 10.1016/j.nbd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 122.Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Human molecular genetics. 2012;21(16):3587–3603. doi: 10.1093/hmg/dds190. [DOI] [PubMed] [Google Scholar]
- 123.Driver JA, Logroscino G, Gaziano JM, Kurth T. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology. 2009;72(5):432–438. doi: 10.1212/01.wnl.0000341769.50075.bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Baumann CR. Epidemiology, diagnosis and differential diagnosis in Parkinson’s disease tremor. Parkinsonism & related disorders. 2012;18(Suppl 1):S90–92. doi: 10.1016/S1353-8020(11)70029-3. [DOI] [PubMed] [Google Scholar]
- 125.Martinez-Ramirez D, Almeida L, Giugni JC, Ahmed B, Higuchi MA, Little CS, Chapman JP, Mignacca C, Wagle Shukla A, Hess CW, Hegland KW, Okun MS. Rate of aspiration pneumonia in hospitalized Parkinson’s disease patients: a cross-sectional study. BMC neurology. 2015;15:104. doi: 10.1186/s12883-015-0362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tjaden K. Speech and Swallowing in Parkinson’s Disease. Topics in geriatric rehabilitation. 2008;24(2):115–126. doi: 10.1097/01.TGR.0000318899.87690.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Iwasaki S, Narabayashi Y, Hamaguchi K, Iwasaki A, Takakusagi M. Cause of death among patients with Parkinson’s disease: a rare mortality due to cerebral haemorrhage. Journal of neurology. 1990;237(2):77–79. doi: 10.1007/BF00314665. [DOI] [PubMed] [Google Scholar]
- 128.Lewitt PA. Levodopa for the treatment of Parkinson’s disease. The New England journal of medicine. 2008;359(23):2468–2476. doi: 10.1056/NEJMct0800326. [DOI] [PubMed] [Google Scholar]
- 129.Arduino DM, Esteves AR, Cortes L, Silva DF, Patel B, Grazina M, Swerdlow RH, Oliveira CR, Cardoso SM. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Human molecular genetics. 2012;21(21):4680–4702. doi: 10.1093/hmg/dds309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. The Journal of biological chemistry. 2008;283(14):9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Elkon H, Don J, Melamed E, Ziv I, Shirvan A, Offen D. Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. Journal of molecular neuroscience: MN. 2002;18(3):229–238. doi: 10.1385/JMN:18:3:229. [DOI] [PubMed] [Google Scholar]
- 132.Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochimica et biophysica acta. 2010;1802(1):29–44. doi: 10.1016/j.bbadis.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 133.Lee KK, Boelsterli UA. Bypassing the compromised mitochondrial electron transport with methylene blue alleviates efavirenz/isoniazid-induced oxidant stress and mitochondria-mediated cell death in mouse hepatocytes. Redox biology. 2014;2:599–609. doi: 10.1016/j.redox.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Todorovic M, Wood SA, Mellick GD. Nrf2: a modulator of Parkinson’s disease? J Neural Transm (Vienna) 2016;123(6):611–619. doi: 10.1007/s00702-016-1563-0. [DOI] [PubMed] [Google Scholar]
- 135.Tretter L, Horvath G, Holgyesi A, Essek F, Adam-Vizi V. Enhanced hydrogen peroxide generation accompanies the beneficial bioenergetic effects of methylene blue in isolated brain mitochondria. Free radical biology & medicine. 2014;77:317–330. doi: 10.1016/j.freeradbiomed.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 136.Delport A, Harvey BH, Petzer A, Petzer JP. Azure B and a synthetic structural analogue of methylene blue, ethylthioninium chloride, present with antidepressant-like properties. Life sciences. 2014;117(2):56–66. doi: 10.1016/j.lfs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 137.Yonutas HM, Vekaria HJ, Sullivan PG. Mitochondrial specific therapeutic targets following brain injury. Brain research. 2016;1640(Pt A):77–93. doi: 10.1016/j.brainres.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 138.Haddad SH, Arabi YM. Critical care management of severe traumatic brain injury in adults. Scandinavian journal of trauma, resuscitation and emergency medicine. 2012;20:12. doi: 10.1186/1757-7241-20-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chesnut RM. Secondary brain insults after head injury: clinical perspectives. New Horiz. 1995;3(3):366–375. [PubMed] [Google Scholar]
- 140.Kimbler DE, Murphy M, Dhandapani KM. Concussion and the adolescent athlete. The Journal of neuroscience nursing: journal of the American Association of Neuroscience Nurses. 2011;43(6):286–290. doi: 10.1097/JNN.0b013e31823858a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380(9847):1088–1098. doi: 10.1016/S0140-6736(12)60864-2. [DOI] [PubMed] [Google Scholar]
- 142.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. British journal of anaesthesia. 2007;99(1):4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 143.Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. The American journal of the medical sciences. 2015;350(2):132–138. doi: 10.1097/MAJ.0000000000000506. [DOI] [PubMed] [Google Scholar]
- 144.Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatric disease and treatment. 2015;11:97–106. doi: 10.2147/NDT.S65815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Maxwell WL. Development of Concepts in the Pathology of Traumatic Axonal and Traumatic Brain Injury. In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): 2015. (Frontiers in Neuroengineering). [Google Scholar]
- 146.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Molecular neurobiology. 2015;51(3):966–979. doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14(2):215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ahmed AI, Bullock MR, Dietrich WD. Hypothermia in Traumatic Brain Injury. Neurosurgery clinics of North America. 2016;27(4):489–497. doi: 10.1016/j.nec.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 149.Bennett MH, Trytko B, Jonker B. Hyperbaric oxygen therapy for the adjunctive treatment of traumatic brain injury. The Cochrane database of systematic reviews. 2012;12:CD004609. doi: 10.1002/14651858.CD004609.pub3. [DOI] [PubMed] [Google Scholar]
- 150.Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. BioMed research international. 2014;2014:723060. doi: 10.1155/2014/723060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suliman NA, Mat Taib CN, Mohd Moklas MA, Adenan MI, Hidayat Baharuldin MT, Basir R. Establishing Natural Nootropics: Recent Molecular Enhancement Influenced by Natural Nootropic. Evidence-based complementary and alternative medicine: eCAM. 2016;2016:4391375. doi: 10.1155/2016/4391375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Camfield DA, Stough C, Farrimond J, Scholey AB. Acute effects of tea constituents L-theanine, caffeine, and epigallocatechin gallate on cognitive function and mood: a systematic review and meta-analysis. Nutrition reviews. 2014;72(8):507–522. doi: 10.1111/nure.12120. [DOI] [PubMed] [Google Scholar]
- 153.Varga MD. Adderall abuse on college campuses: a comprehensive literature review. Journal of evidence-based social work. 2012;9(3):293–313. doi: 10.1080/15433714.2010.525402. [DOI] [PubMed] [Google Scholar]
- 154.Gonzalez-Lima F, Barksdale BR, Rojas JC. Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochemical pharmacology. 2014;88(4):584–593. doi: 10.1016/j.bcp.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 155.Rodriguez P, Zhou W, Barrett DW, Altmeyer W, Gutierrez JE, Li J, Lancaster JL, Gonzalez-Lima F, Duong TQ. Multimodal Randomized Functional MR Imaging of the Effects of Methylene Blue in the Human Brain. Radiology. 2016;281(2):516–526. doi: 10.1148/radiol.2016152893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Barrett DW, Gonzalez-Lima F. Transcranial infrared laser stimulation produces beneficial cognitive and emotional effects in humans. Neuroscience. 2013;230:13–23. doi: 10.1016/j.neuroscience.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 157.Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017 doi: 10.1111/jnc.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Grimm A, Friedland K, Eckert A. Mitochondrial dysfunction: the missing link between aging and sporadic Alzheimer’s disease. Biogerontology. 2016;17(2):281–296. doi: 10.1007/s10522-015-9618-4. [DOI] [PubMed] [Google Scholar]
- 159.Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res. 2011;8(4):393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Voloboueva LA, Giffard RG. Inflammation, mitochondria, and the inhibition of adult neurogenesis. Journal of neuroscience research. 2011;89(12):1989–1996. doi: 10.1002/jnr.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132(4):645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 162.Kernie SG, Parent JM. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Dis. 2010;37(2):267–274. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang RL, Zhang ZG, Zhang L, Chopp M. Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience. 2001;105(1):33–41. doi: 10.1016/s0306-4522(01)00117-8. [DOI] [PubMed] [Google Scholar]
- 164.Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18(19):7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nature medicine. 2002;8(9):963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 166.Bettio LEB, Rajendran L, Gil-Mohapel J. The effects of aging in the hippocampus and cognitive decline. Neurosci Biobehav Rev. 2017;79:66–86. doi: 10.1016/j.neubiorev.2017.04.030. [DOI] [PubMed] [Google Scholar]
- 167.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta neuropathologica. 2003;106(6):518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 168.Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer research. 2003;63(14):4021–4027. [PubMed] [Google Scholar]
- 169.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nature medicine. 2002;8(12):1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 170.Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1999;19(1):87–98. doi: 10.1097/00004647-199901000-00010. [DOI] [PubMed] [Google Scholar]
- 171.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 172.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302(5651):1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 173.Torroglosa A, Murillo-Carretero M, Romero-Grimaldi C, Matarredona ER, Campos-Caro A, Estrada C. Nitric oxide decreases subventricular zone stem cell proliferation by inhibition of epidermal growth factor receptor and phosphoinositide-3-kinase/Akt pathway. Stem Cells. 2007;25(1):88–97. doi: 10.1634/stemcells.2006-0131. [DOI] [PubMed] [Google Scholar]
- 174.Fike JR, Rola R, Limoli CL. Radiation response of neural precursor cells. Neurosurgery clinics of North America. 2007;18(1):115–127. doi: 10.1016/j.nec.2006.10.010. x. [DOI] [PubMed] [Google Scholar]
- 175.Cordeau-Lossouarn L, Vayssiere JL, Larcher JC, Gros F, Croizat B. Mitochondrial maturation during neuronal differentiation in vivo and in vitro. Biology of the cell. 1991;71(1-2):57–65. doi: 10.1016/0248-4900(91)90051-n. [DOI] [PubMed] [Google Scholar]
- 176.Kirby DM, Rennie KJ, Smulders-Srinivasan TK, Acin-Perez R, Whittington M, Enriquez JA, Trevelyan AJ, Turnbull DM, Lightowlers RN. Transmitochondrial embryonic stem cells containing pathogenic mtDNA mutations are compromised in neuronal differentiation. Cell proliferation. 2009;42(4):413–424. doi: 10.1111/j.1365-2184.2009.00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Papa S, Petruzzella V, Scacco S, Vergari R, Panelli D, Tamborra R, Corsi P, Picciariello M, Lambo R, Bertini E, Santorelli FM. Respiratory complex I in brain development and genetic disease. Neurochemical research. 2004;29(3):547–560. doi: 10.1023/b:nere.0000014825.42365.16. [DOI] [PubMed] [Google Scholar]
- 178.Wong A, Cavelier L, Collins-Schramm HE, Seldin MF, McGrogan M, Savontaus ML, Cortopassi GA. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Human molecular genetics. 2002;11(4):431–438. doi: 10.1093/hmg/11.4.431. [DOI] [PubMed] [Google Scholar]
- 179.Xie L, Choudhury GR, Wang J, Park Y, Liu R, Yuan F, Zhang CL, Yorio T, Jin K, Yang SH. Methylene blue promotes quiescence of rat neural progenitor cells. Frontiers in cellular neuroscience. 2014;8:315. doi: 10.3389/fncel.2014.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.van der Ven AT, Pape JC, Hermann D, Schloesser R, Genius J, Fischer N, Mossner R, Scherbaum N, Wiltfang J, Rujescu D, Benninghoff J. Methylene Blue (Tetramethylthionine Chloride) Influences the Mobility of Adult Neural Stem Cells: A Potentially Novel Therapeutic Mechanism of a Therapeutic Approach in the Treatment of Alzheimer’s Disease. J Alzheimers Dis. 2017;57(2):531–540. doi: 10.3233/JAD-160755. [DOI] [PubMed] [Google Scholar]
- 181.Crooks J. Haemolytic jaundice in a neonate after intra-amniotic injection of methylene blue. Archives of disease in childhood. 1982;57(11):872–873. doi: 10.1136/adc.57.11.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Porat R, Gilbert S, Magilner D. Methylene blue-induced phototoxicity: an unrecognized complication. Pediatrics. 1996;97(5):717–721. [PubMed] [Google Scholar]
- 183.Spahr RC, Salsburey DJ, Krissberg A, Prin W. Intraamniotic injection of methylene blue leading to methemoglobinemia in one of twins. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics. 1980;17(5):477–478. doi: 10.1002/j.1879-3479.1980.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 184.Wolvetang T, Janse R, Ter Horst M. Serotonin Syndrome After Methylene Blue Administration During Cardiac Surgery: A Case Report and Review. Journal of cardiothoracic and vascular anesthesia. 2016;30(4):1042–1045. doi: 10.1053/j.jvca.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 185.Hencken L, To L, Ly N, Morgan JA. Serotonin Syndrome Following Methylene Blue Administration for Vasoplegic Syndrome. Journal of cardiac surgery. 2016;31(4):208–210. doi: 10.1111/jocs.12705. [DOI] [PubMed] [Google Scholar]
- 186.Smith CJ, Wang D, Sgambelluri A, Kramer RS, Gagnon DJ. Serotonin syndrome following methylene blue administration during cardiothoracic surgery. Journal of pharmacy practice. 2015;28(2):207–211. doi: 10.1177/0897190014568389. [DOI] [PubMed] [Google Scholar]



