
Por Alexandra LATYPOVA fundadora de iCardiac Technologies, una empresa basada en tecnología desarrollada por estudiantes y profesores de la Universidad de Rochester. Atiende a 6 de las 10 principales compañías farmacéuticas.

Resumen
El desarrollo apresurado y la aprobación a «acelerada» de inyecciones de ADN y ARNm contra Covid-19 completamente nuevas que se han impuesto a las personas en todo el mundo ha resultado hasta hoy en millones de lesiones reportadas y miles de muertes según bases de datos de salud pública como VAERS (EE. UU.) , EudraVigilance (UE), Yellow Card (Reino Unido) y otros. Este artículo revisa algunos de los documentos disponibles públicamente sobre el programa de desarrollo no clínico de Pfizer y señala sus deficiencias, omisiones y lagunas, que eran muy obvias, pero que nunca fueron cuestionadas por los reguladores u otras autoridades sanitarias. La naturaleza superficial de todo el programa preclínico se puede resumir como «no encontramos ninguna señal de seguridad porque no la buscamos«. La omisión de estudios de seguridad que se consideran estándar o incluso obligatorios, y la deshonestidad científica en los estudios que se realizaron es tan evidente y flagrante que no se puede atribuir a la incompetencia de los fabricantes y reguladores. Más bien, debe plantearse la cuestión como negligencia deliberada.
El enfoque de esta revisión es el alcance y la adecuación del programa de evaluación no clínica para una nueva inyección de terapia génica, combinado con una breve discusión de los marcos regulatorios relevantes. No se profundizó en la revisión de los resultados de estudios específicos. El objetivo es ilustrar el colapso total del proceso regular de desarrollo y aprobación de medicamentos, que antes era riguroso y ético, así como la negligencia impactante por parte de las agencias reguladoras que se supone deben mantenerse honestos. Esta revisión, demuestra los siguientes hallazgos:
- El programa de Pfizer no incluyó una prueba completa de todos los componentes del producto final aprobado de la inyección mRNA COVID-19. En cambio, los estudios incluidos en el paquete de documentos presentado a la FDA emplearon varias variantes y análogos del producto, cuya comparabilidad con la inyección COVID-19 real no se demostró ni evaluó. Por lo tanto, no se puede realizar una evaluación exhaustiva de la seguridad del producto sobre la base de estos estudios.
- Un determinante clave de la toxicidad de una droga es su distribución dentro del cuerpo. Sin embargo, con el ingrediente activo de ARNm de la inyección COVID-19 de Pfizer, ¡este aspecto crucial nunca se estudió!
- Pfizer afirmó la ausencia de potencial para el «aumento de la enfermedad provocada por la inyección» con base en estudios de una especie animal que no se enferma por el SARS-CoV-2.
- Los CDC, la FDA y Pfizer mintieron acerca de que «la vacuna permanece en el lugar de la inyección«; sabían desde el principio que la distribución de la inyección por todo el cuerpo era de esperar.
- Pfizer se saltó por completo las principales categorías de pruebas de seguridad.
- Pfizer utilizó una interpretación deshonesta de las pautas regulatorias para justificar los atajos que tomó en las pruebas de seguridad de rutina.
- Tanto la FDA como Pfizer conocían las principales toxicidades asociadas con los medicamentos de terapia génica en general y, por lo tanto, no pueden alegar falta de conocimiento anticipado de estos riesgos con el medicamento de terapia génica particular que es la inyección COVID-19 de Pfizer. Esto apunta a un fraude intencional y colusión entre Pfizer y los reguladores, quienes conspiraron para impulsar este peligroso producto no probado en el mercado.
En general, por lo tanto, tanto el fabricante como los reguladores se comportaron de una manera muy deshonesta y conspiraron para impulsar una tecnología y un producto completamente nuevos a millones de personas sin llevar a cabo una sola evaluación de seguridad bien diseñada.
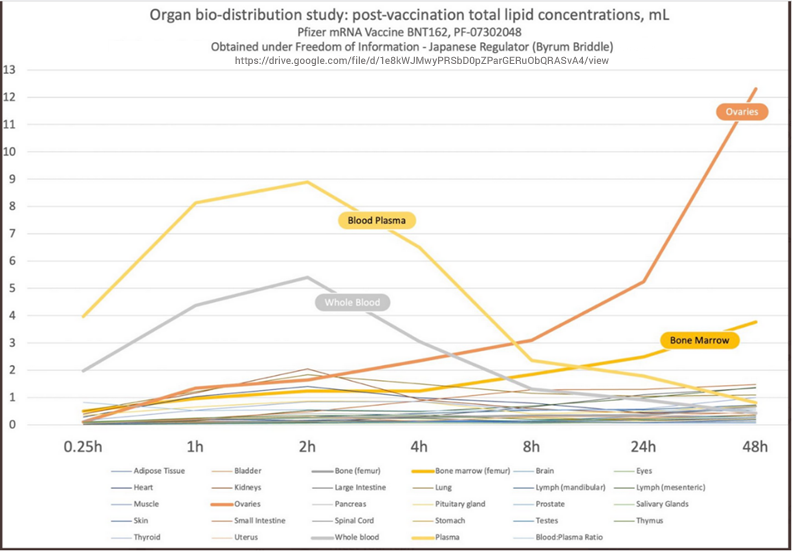
Los japoneses recurrieron a una demanda FOIA (Freedom of Information Act) para obligar a Pfizer a publicar su estudio secreto de biodistribución. La razón por la que Pfizer quería que se mantuviera en secreto es que demostraba que Pfizer mintió al público y a las agencias reguladoras sobre el destino del contenido de la inyección (nanolípidos que contiene el ARNm). Pfizer dijo que permanecía en el sitio de la inyección (el hombro), cuando de hecho su propio estudio revela que se diseminó rápidamente por todo el cuerpo a través del torrente sanguíneo en 48 horas. Tambien queda en evidencia que estos portadores de nanolípidos se acumularon en concentraciones muy altas en varios órganos, incluidos los órganos reproductivos de hombres y mujeres, el corazón, el hígado, la médula ósea y el bazo (un órgano inmunológico importante). La concentración más alta estaba en los ovarios y la médula ósea. Estos portadores de nanolípidos también se depositaron en el cerebro.
https://www.docdroid.net/xq0Z8B0/pfizer-report-japanese-government-pdf
Roxana Bruno, Licenciada en Bioquímica, Dra. en Inmunología, Universidad Autónoma de Barcelona, advirtiendo mediados de 2021.
La proteina Spike o pico que genera la inyección K0 BIT es nociva al cuerpo humano. https://www.bitchute.com/video/3nvMvK4zQ17d/
min. 3 Como funcionan estas inyecciones
min. 11 Presentacion de estudio sobre inseguridad de las inyecciones
min. 14 Efectos adversos
min. 15 ADE Variantes
min. 16 Estudios con animales variantes
min. 18 Vacunacion anual constante
min. 19:25 Trombosis
min. 21:44 Trombosis Cerebrales
min. 22 Trombositopenia
min. 23:40 Factor plaquetario 4
min. 24:15 Trombosis inducidas por vacunas: Trombosis trombocitopénica inmune
inducida por la vacuna (VIPIT por sus siglas en inglés).
min. 28:40 Trombocitopenia, EMA clasifica como efecto secundario comun
es decir, 1 de cada 100 vacunado a 1 de cada 10 Trombocitopenia
con la inyección de Astra zeneca
min. 32:50 Reportar Efectos Adversos
min. 33:25 La proteina espiga o Spike de la inyección es toxica al ser humano
1. Antecedentes
El proceso de investigación y desarrollo, I+D farmacéutico está fuertemente regulado y dividido en múltiples fases, con el objetivo de:
- eliminar el riesgo de nuevos medicamentos,
- reducir el potencial de daño a los seres humanos y
- garantizar una comprensión suficiente de los riesgos y beneficios de un nuevo medicamento en cada paso.
La etapa inicial de este proceso son los estudios preclínicos, en los que se prueba un fármaco o producto biológico en líneas celulares, animales pequeños (p. ej., ratones y ratas) y animales más grandes (p. ej., monos). El objetivo de los estudios preclínicos es demostrar que el nuevo medicamento tiene el modo de acción previsto, así como caracterizar la seguridad y la eficacia a un nivel suficiente para decidir si se justifican los estudios clínicos. Para las vacunas tradicionales, la fase no clínica es la única fase de desarrollo en la que se evalúan formalmente la seguridad y la toxicidad.
La Administración de Drogas y Alimentos de los EE. UU. (FDA) proporciona pautas para las diferentes fases de desarrollo, que se describen en las publicaciones de Orientación para la industria de la FDA [ 1 ] y se coordinan globalmente a través de la Conferencia Internacional sobre Armonización (International Conference on Harmonization , ICH). Se han emitido conjuntos separados de directrices para medicamentos convencionales (moléculas pequeñas) y productos biológicos, incluidas las vacunas. Sin embargo, para plataformas más complejas, como combinaciones de fármaco-dispositivo o fármaco-biológico, será aplicable más de una directriz, dependiendo de la composición del producto terminado. Desde 2013, la FDA ha estado publicando pautas que se refieren específicamente a las plataformas de terapia génica. La guía más reciente de la FDA sobre pruebas de fase temprana de productos de terapia génica se publicó en 2015 [2] . Los aspectos relevantes de esta guía y la posición de la FDA sobre esta clase de medicamentos se analizan al final de este documento (ver en la Sección 2.7 ), que demostrará lo que la FDA y los fabricantes sabían sobre los riesgos asociados con la clase antes de 2020.
El alcance exacto del programa de desarrollo de cada nuevo medicamento se negocia en una serie de reuniones entre el fabricante y la FDA. En general, cuanto más novedoso sea el nuevo fármaco o entidad biológica, más estrictas y extensas serán las pruebas requeridas, ya que con los nuevos medicamentos los riesgos aún no están bien caracterizados y los datos de seguridad relevantes de la experiencia previa con productos similares son escasos. Es necesario evaluar numerosos riesgos desconocidos de daño potencial para los pacientes, y cualquier riesgo que se identifique en las primeras pruebas debe caracterizarse minuciosamente en estudios posteriores, de modo que al final se pueda realizar una evaluación de riesgo/beneficio bien informada.
Por necesidad, un fármaco o un producto biológico debe considerarse peligroso hasta que se demuestre su seguridad. Afirmar que algo es seguro por razones puramente teóricas o porque “todas las vacunas son seguras” no es aceptable desde el punto de vista científico o ético. Además, cabe señalar que históricamente la FDA no ha permitido la prueba de diferentes versiones de un producto candidato bajo la misma solicitud de nuevo fármaco en investigación (IND).
Una estrategia de pruebas no clínicas bien diseñada incluirá la caracterización del producto y sus componentes en las siguientes categorías generales de investigación (cada una de las cuales se analiza más adelante en este artículo):
- la farmacocinética, que se refiere a la absorción, distribución y eliminación del fármaco del cuerpo;
- la farmacodinámica, es decir, el mecanismo de acción del fármaco, incluidos los efectos primarios y secundarios (fuera del objetivo);
- farmacología y toxicología de seguridad, incluida la caracterización de riesgos para las principales clases de órganos: sistema cardiovascular y nervioso central, hígado, riñones y sangre, así como otros sistemas de órganos seleccionados en función de los efectos conocidos o previstos de la clase de producto o sus componentes;
- genotoxicidad, es decir, la propensión del fármaco a causar daño al material genético (ADN);
- carcinogenicidad (propensión a causar cáncer);
- toxicología reproductiva, que se refiere a la toxicidad para los órganos reproductivos o para el feto en desarrollo. Esto debe evaluarse antes de que el producto pueda administrarse a personas con potencial reproductivo;
- otro tipo de estudios diseñados para caracterizar el riesgo en base a las señales de seguridad identificadas en cualquiera de los estudios iniciales anteriores.
Es importante tener en cuenta que, si bien las agencias globales como la Organización Mundial de la Salud pueden proporcionar opiniones técnicas o científicas a través de recomendaciones publicadas, en los Estados Unidos la autoridad única para regular el desarrollo de medicamentos/biológicos y aprobar nuevos productos recae en la FDA.
2. Programa no clínico de Pfizer para su inyección de ARNm Covid-19
Recientemente, algunos de los documentos utilizados por la FDA para aprobar la inyección Covid-19 basada en ARNm de Pfizer se obtuvieron a través de solicitudes y demandas de Freedom o Information FOIA, superando las mociones de la FDA y Pfizer de mantener esta información en secreto durante 75 años. Judicial Watch obtuvo del Departamento de Salud y Servicios Humanos [ 3 ] un paquete de estos documentos sobre ensayos preclínicos, que suman 466 páginas .
2.1. El programa de Pfizer no incluía una prueba completa de principio a fin de todos los componentes del producto final aprobado.
Los estudios incluidos en el paquete de aprobación fueron para una variedad de versiones del producto sin evaluaciones de comparabilidad; por lo tanto, no se puede realizar una evaluación exhaustiva de la seguridad del producto. La página 6 del “Módulo no clínico” contenido en los documentos de la FOIA [ 3 ] establece lo siguiente (énfasis añadido): BNT162b2 (número de código de BioNTech BNT162, número de código de Pfizer PF-07302048) es una inyección en investigación destinada a prevenir el COVID-19, que es causado por el SARS-CoV-2. BNT162b2 es un ARNm modificado con nucleósidos (ARNmod) que expresa S [proteína pico o Spike] de longitud completa con dos mutaciones de prolina (P2) para bloquear la proteína transmembrana en una conformación de prefusión antigénicamente óptima … La inyección está formulada en nanopartículas lipídicas (LNP). El LNP está compuesto por 4 lípidos: ALC-0315, ALC-0159, DSPC y colesterol . Otros excipientes en la formulación incluyen sacarosa, NaCl, KCl, Na 2 HPO 4 y KH 2 PO 4 . La dosis seleccionada para BNT162b2, con eficacia demostrada en la evaluación clínica de Fase 2/3 y prevista para uso comercial, es de 30 µg administrados IM en dos dosis administradas con 21 días de diferencia.
Está claro a partir de la descripción del producto anterior que esta plataforma completamente novedosa consta de nuevos componentes biológicos/genéticos y químicos patentados dentro de una estructura de “carga útil más vehículo de entrega”. Siempre que los productos complejos contengan combinaciones de medicamentos y productos biológicos, o productos biológicos y nuevos vehículos de administración como ocurre con el producto de Pfizer, el fabricante debe evaluar la seguridad de todos los componentes por separado y también en la versión ensamblada final que está destinada a las fases humanas de desarrollo [ 4 ] .
La misma página del documento FOIA explica además: En estudios no clínicos, se probaron dos variantes de BNT162b2; designadas como «variante 8» y «variante 9» (V8 y V9, respectivamente). Las variantes difieren solo en sus secuencias de optimización de codones que están diseñadas para mejorar la expresión de antígenos; de lo contrario, las secuencias de aminoácidos de los antígenos codificados son idénticas. Solo BNT162b2 (V9) ha sido evaluado en la clínica, actualmente está autorizado bajo EUA y es el sujeto de esta solicitud BLA.
La declaración resaltada arriba es falsa, al menos con respecto a las pruebas clínicas. La revisión de los estudios clínicos publicados por la FOIA reveló que se incluyeron al menos 4 variantes diferentes del ingrediente activo en la única solicitud de nuevo fármaco en investigación de Pfizer IND n.º 19736:
- Proteína pico o Spike de SARS-CoV-2; MP: No veo ensayos clínicos sobre la proteína en sí mencionada en el paquete; el único lugar donde encuentro la frase «Proteína pico de SARS-CoV-2» es en las referencias bibliográficas.
- BNT162a1: ARNm no modificado (ARNu; variante RBL063.3);
- BNT162b1: ARNm modificado con metilpseudouridina (ARNmod; variante RBP020.3);
- BNT162b2: ARN modificado con metilpseudouridina (modRNA; variante RBP020.2);
- BNT162c2: ARNm no modificado autoamplificador (ARNsa; variante RBS004.2)
Cada tipo de ARNm puede administrarse utilizando las mismas nanopartículas lipídicas compuestas de ALC-0315, ALC-0159, diestearoil-fosfatidilcolina (DSPC) y colesterol [ 5 ] . Según el Folleto del investigador emitido por BioNTech, que la FOIA obtuvo del regulador australiano (TGA), varias versiones de RNA, modRNA y saRNA se estaban utilizando en múltiples estudios clínicos en varios países hasta agosto de 2020 [ 6 ] . Además, BNT162b1 en lugar de la variante b2 fue el artículo de prueba utilizado en el ensayo clínico de Fase I de Pfizer [ 7 ]. Del mismo modo, es evidente a partir del protocolo del ensayo clínico de Fase 1/2/3 y sus enmiendas que se agregaron nuevas versiones no mencionadas en la solicitud IND anterior sin explicación de su composición o ninguna prueba nueva (por ejemplo, una variante específica de Sudáfrica del la vacuna se agregó al calendario del protocolo en 2021).
Si bien el uso de múltiples versiones de un producto en las primeras etapas de desarrollo suele ser inevitable, cada entidad química o biológica se considera legalmente distinta a los efectos de la aprobación del producto. Por lo tanto, los estudios realizados con versiones del producto que no se ajustan a la especificación exacta de la versión final pueden servir solo como información de respaldo para la aprobación de esta última, pero nunca deben considerarse pruebas definitivas y suficientes para afirmaciones de seguridad o eficacia relativa al producto final.
En septiembre de 2021, la FDA emitió un borrador de guía titulado Estudio de versiones múltiples de un producto de terapia celular o génica en un ensayo clínico de fase inicial [ 8 ], que establece que cada versión del producto requiere una aplicación IND separada. Sin embargo, una nota a pie de página en esta guía exime a las “vacunas destinadas a prevenir enfermedades infecciosas” de este requisito. No se da ninguna explicación de por qué se hace esta exención, y no existe ninguna base científica o legal concebible para esta exención, aparte de que la FDA ya había permitido arbitrariamente esta desviación sin precedentes del estándar regulatorio y luego necesitaba cubrir sus huellas. De hecho, podría decirse que esta «excepción» regulatoria ni siquiera se aplica a la «vacuna» COVID-19 de Pfizer, ya que el producto no previene la infección o la transmisión de la enfermedad. ¿Es la intención de prevenir la enfermedad por sí sola una condición suficiente? Después de todo, cada medicamento nuevo tiene la intención de hacer algo como prevenir una enfermedad, pero solo unos pocos lo logran con éxito.
Pfizer afirma que la farmacología primaria, la distribución, el metabolismo, la seguridad y la inmunogenicidad de BNT162b2 se estudiaron in vitro e in vivo en ratones, ratas y monos rhesus, así como en varios experimentos de ensayos de cultivos celulares. En total, se incluyeron 18 estudios en el paquete no clínico, de los cuales 7 fueron para la variante de secuencia de nucleótidos V9; estos incluyeron un estudio que no cumplió con las normas de «buenas prácticas de laboratorio» (BPL) [ 9 ] por lo tanto, no debería haberse considerado aceptable para la aprobación reglamentaria y el etiquetado. Seis de los estudios incluidos se referían a dos de los cuatro excipientes lipídicos, ALC-0315 y ALC-0159. No se estudiaron los otros lípidos incluidos en la Plataforma de nanopartículas lipídicas (LNP), a saber, diestearoil-fosfatidilcolina (DSPC) y colesterol. Pfizer y los reguladores argumentaron en otro lugar que el DSPC y el colesterol «se producen de forma natural», lo que en general es cierto. Sin embargo, ninguno ocurre en la naturaleza como parte de la formulación exacta de nanopartículas de lípidos que se usa en el producto de Pfizer. De hecho, algunas publicaciones de Moderna se refieren a análogos de colesterol, que fueron sustituidos por colesterol para mejorar la penetración en la célula [ 10 ]. Los documentos de Pfizer no explican qué forma de colesterol se usa y cómo se formula. No se proporcionan pruebas de biocompatibilidad, biosimilitud o toxicidad.
De los seis estudios sobre los excipientes lipídicos, cuatro fueron para formulaciones lipídicas «comparables a LNP en BNT162b2» y dos estudios no se ajustaron a las buenas prácticas de laboratorio (BPL). No se explica en ninguna parte del documento cómo estas otras formulaciones diferían de la formulación final de la LNP incluida en el producto aprobado, y cómo se determinó más allá de la afirmación de Pfizer de que eran realmente comparables. Por lo tanto, solo 9 de los 18 estudios en este paquete están directamente relacionados con el producto licenciado o con componentes especificados con precisión del producto final.
2.2. Si bien se realizaron algunos estudios de toxicidad limitados, ¡nunca se estudió la farmacocinética completa del ARNm del ingrediente activo!
En los estudios diseñados para evaluar si la inyección permanece cerca del lugar de la inyección o viaja por todo el cuerpo, Pfizer no utilizó el artículo de prueba representativo de la inyección comercial, que se llama BNT162b2 y contiene ARNm modificado con metil-pseudouridina que codifica para la inyección de longitud completa de la proteína pico o espiga con dos mutaciones de prolina (P2). En cambio, Pfizer estudió la biodistribución mediante la administración de “luciferasa codificante de ARN modificada formulada en LNP comparable a BNT162b2 con trazas de [ 3 H]-CHE [colesteril hexadecil éter] como etiqueta no difusible” [ 3 , p. 10] a ratones y ratas, es decir, un ARNm «sustituto» que codifica la enzima luciferasa en lugar de la proteína de punta del SARS-CoV-2.
Los resultados de ese estudio se resumen en la Figura 1 a continuación. Pfizer y la FDA simplemente asumen que la inyección real mostrará el mismo patrón de distribución, porque su composición lipídica es idéntica [ 3 , p. 43] (énfasis añadido): Es probable que la distribución al hígado esté mediada por LNP que ingresan al torrente sanguíneo. La expresión de luciferasa en los sitios de inyección cayó a los niveles de fondo después de 9 días. El estudio de toxicidad de dosis repetidas en ratas no mostró evidencia de daño hepático (Sección 2.4.4.3). Se espera que la biodistribución del antígeno codificado por el componente de ARN de BNT162b2 dependa de la distribución de LNP y los resultados presentados deberían ser representativos de la plataforma de ARN de la inyección , ya que el ARN mod codificante de luciferasa formulado por LNP tenía la misma composición lipídica.
Sin embargo, esta afirmación no está respaldada por ningún dato y es científicamente insostenible. En realidad, los estudios que emplean la proteína luciferasa presumiblemente inerte probablemente representan el «mejor de los casos». Incluso esta inyección modelo viaja por todo el cuerpo, y el estudio en cuestión demuestra también la expresión de luciferasa en el bazo y el hígado. Es muy posible que empeore con el ARNm que codifica la espiga: la fuga vascular inducida por la proteína espiga expresada podría muy bien aumentar la penetración de las nanopartículas de la vacuna en los tejidos de otros órganos, en particular también en el cerebro [ 11 – 13 ] .
Sin embargo, la afirmación de Pfizer no solo es científicamente deshonesta (están utilizando una hipótesis no probada en lugar de una prueba), sino que esta declaración es clave para la aprobación de toda la «plataforma de vacunas» fraudulenta: Pfizer, con la connivencia de la FDA, quiere que el público y los médicos profesionales a creer que el vehículo de entrega (LNP) es el «producto», mientras que la carga útil que se entrega es irrelevante y puede sustituirse por sustitutos arbitrarios. Esto va en contra de todas las prácticas regulatorias previamente establecidas en I+D farmacéutica, el método científico e incluso el sentido común. ¡Es como decir que un camión cargado de alimentos y un camión cargado de explosivos son lo mismo!
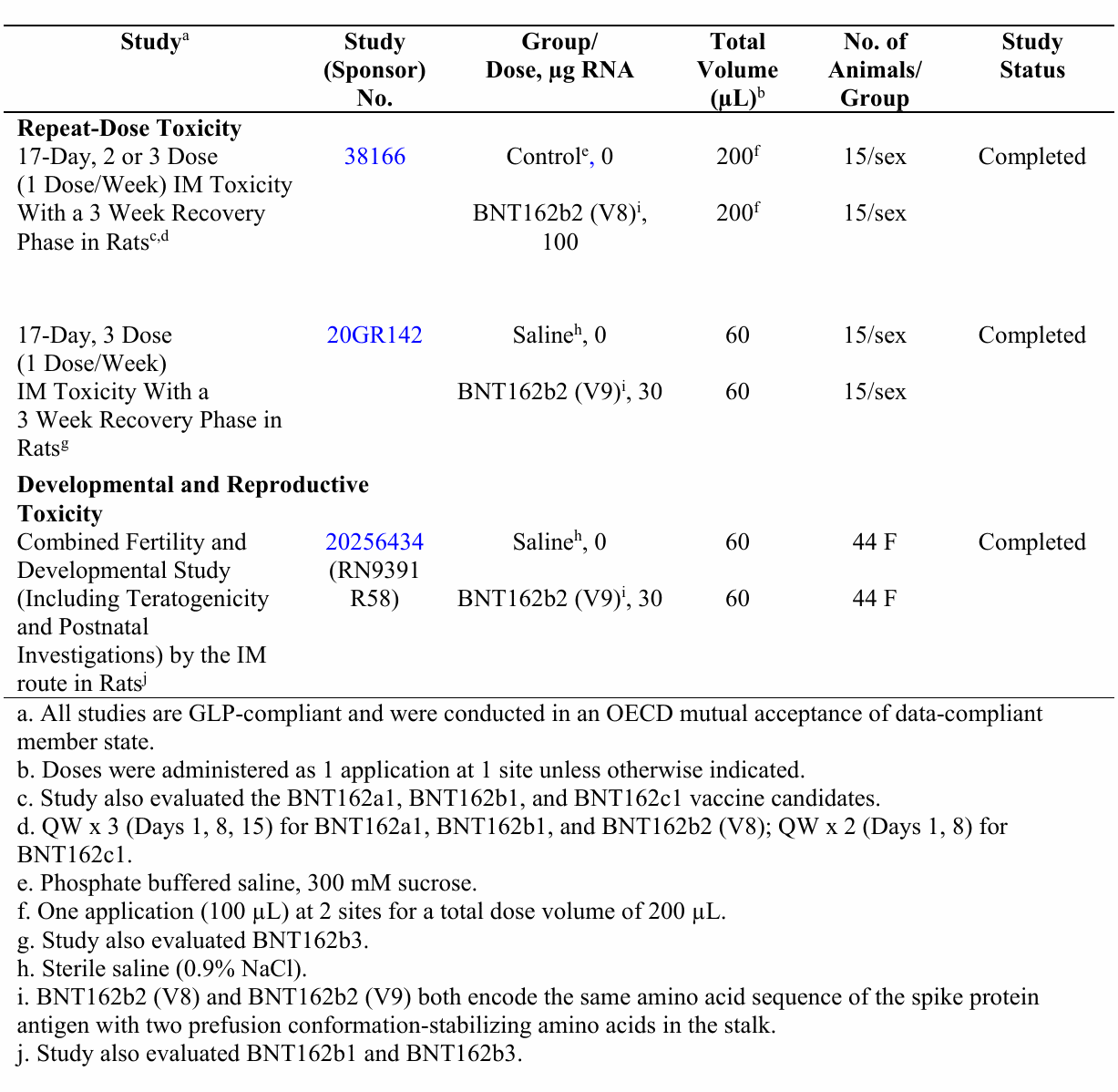
Si bien se realizaron algunos estudios de toxicología, el programa general fue extremadamente limitado. Estos estudios se resumen en la Tabla 1 a continuación, que se tomó del paquete de documentos no clínicos de Pfizer. De acuerdo con la tabla, hubo un total de solo tres estudios en ratas, de los cuales solo 2 eran estudios de toxicidad de dosis repetidas que cumplían con las buenas prácticas de laboratorio (BPL), y uno de los dos incluía una versión diferente del producto (V8) que difiere de BNT162b2 ( V9), el candidato presentado para obtener la licencia en “presencia de codones optimizados para mejorar la expresión de antígenos”. No hay datos disponibles sobre la comparabilidad de las diferentes versiones de ARNm. Pfizer simplemente afirma que los cambios son para «optimización» y «no se espera» que influyan en la seguridad.
A pesar de la afirmación en la nota al pie de la tabla (a), no los tres estudios se realizaron de conformidad con las buenas prácticas de laboratorio. El estudio de toxicidad para el desarrollo y la reproducción (DART) en ratas Wistar Han no cumplió con las buenas prácticas de laboratorio (BPL) (esto se revela en el texto del documento). El estudio de toxicidad para la reproducción que no cumple con las BPL solo puede considerarse exploratorio. Además, las ratas macho del estudio no se trataron con el producto final de Pfizer y, por lo tanto, no se evaluó el impacto de la vacunación en la fertilidad de los machos.
Los resultados de estos estudios solo se analizan brevemente en un resumen redactado de forma ambigua que puede significar que no se detectaron muertes ni anomalías, o que se detectaron pero los investigadores consideraron que no estaban relacionadas con la inyección. No hay forma de verificar de forma independiente estas afirmaciones, ya que el informe completo del estudio no está disponible. El estudio también confirmó que los anticuerpos (y, por lo tanto, probablemente también las proteínas pico) pasan de la madre a la descendencia, un detalle muy importante que nunca se mencionó en los anuncios de los CDC de esta inyección como «segura» para las mujeres embarazadas.
El único estudio de toxicología que se realizó con la versión correcta de la inyección candidata y de acuerdo con las BPL fue el estudio de toxicidad de dosis repetidas # 20GR142. El informe completo de este estudio no está disponible. La propia descripción de los resultados del fabricante indica que los animales experimentaron pérdida de apetito y pérdida de peso, fiebre, patología clínica y cambios en los parámetros de laboratorio consistentes con inflamación, y no todos los cambios se resolvieron cuando finalizó el estudio. Los animales tratados tenían bazos agrandados (1,5 veces) y ganglios linfáticos. Se observaron los hallazgos patológicos en el hígado, el bazo, la médula ósea y los ganglios linfáticos, pero no se describieron en detalle, y el fabricante los descartó por considerarlos no ser significativos. No hay forma de evaluar de forma independiente estos hallazgos.
Dado que tanto el fabricante como la FDA están luchando para mantener en secreto los datos de toxicología y no los han revelado completamente en esta respuesta a la FOIA, solo podemos concluir que los hallazgos en los estudios con animales fueron graves.
Habría sido posible evaluar la expresión (y los efectos subsiguientes o la ausencia de los mismos) de la proteína pico, o espiga (spike) en varios tejidos de interés. Recientes estudios histopatológicos sobre materiales de autopsia de muertes posteriores a la inyección muestran claramente que la expresión de la proteína espiga con el subsiguiente daño orgánico puede detectarse y estudiarse fácilmente con técnicas estándar, incluso meses después de la inyección más reciente [ 14 ] . Esto habría sido aún más fácil en un entorno experimental.
2.3. Pfizer cita estudios de una especie animal que no se enferma por el SARS-CoV-2 para afirmar la ausencia de «aumento de la enfermedad provocada por la inyección».
La aumento o mejora potencial de la enfermedad, ADE es un riesgo conocido que se ha identificado en numerosos estudios previos en animales con medicamentos de terapia génica. Pfizer y la FDA eran claramente conscientes de este riesgo. Para «probar» que este riesgo no se aplica a su inyección de ARNm, Pfizer se refirió a un estudio de inmunogenicidad (VR-VTR-10671) en monos rhesus. Seis animales recibieron dos inyecciones con 21 días de diferencia, mientras que un grupo de control de tres monos no recibió ninguna inyección. Los monos inoculados produjeron una respuesta de anticuerpos mensurable, y tras el desafío viral produjeron niveles mucho más bajos de ARN viral en los pulmones que el grupo de control.
El paquete de documentos también enfatiza repetidamente que en este estudio muy pequeño «no hubo evidencia de aumento o mejora de la enfermedad provocada por la inyección». Sin embargo, de manera crucial, ninguno de los monos en ninguno de los grupos se enfermó clínicamente. Pfizer lo afirma explícitamente [ 3 , p. 15] (énfasis añadido): Ninguno de los animales desafiados mostró signos clínicos de enfermedad significativa, lo que indica que el modelo de desafío de rhesus macho de 2 a 4 años es principalmente un modelo de infección por SARS-CoV-2, no un modelo de enfermedad COVID-19 .
Parece que para Pfizer, así como para la FDA, que aceptó alegremente esta «evidencia», los estudios de un modelo animal que carece de enfermedad en primer lugar son suficientes para demostrar la falta de enfermedad aumentada o mejorada.
2.4. CDC, FDA y Pfizer mintieron acerca de que «la inyección permanece en el lugar de la inyección».
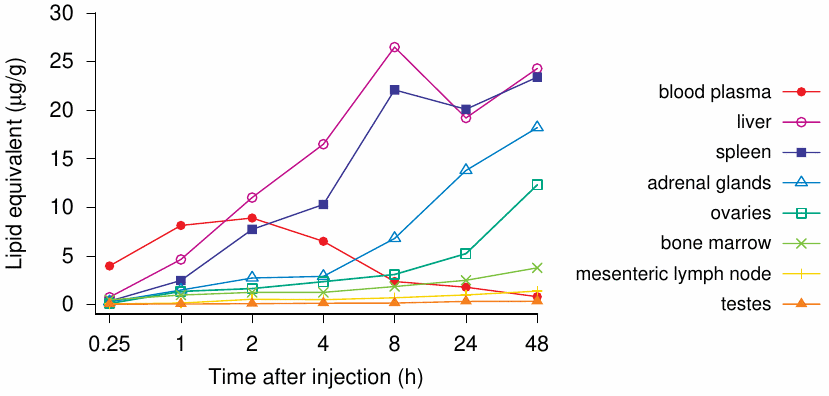
El estudio modelo de biodistribución de la inyección discutido en la Sección 2.2 muestra claramente que la carga útil, sea lo que sea, un sustituto de la luciferasa o un ARNm nunca probado que codifica la proteína pico o espiga (spike), ingresa al torrente sanguíneo y se distribuye por todo el cuerpo (Figura 1 ). Como se ve en el gráfico, hay grandes acumulaciones en las glándulas suprarrenales, el hígado, el bazo, los ovarios y otros órganos, como los ganglios linfáticos y la médula ósea, como se ilustra en la siguiente tabla. De hecho, uno de los estudios en ratas incluidos en el envase prevé que el producto llegue directamente al torrente sanguíneo e incluye la vía de administración intravenosa. Este estudio también se basa en el ARNm sustituto y no en el que codifica la proteína pico o espiga.
Hay motivos para preocuparse por las implicaciones clínicas de estos hallazgos. Sin embargo, dado que el enfoque de este artículo es el alcance del programa no clínico de Pfizer y no una revisión en profundidad de estos estudios, remito a los lectores a un excelente análisis realizado por científicos expertos en este campo [ 15 ]. En el contexto preclínico, debe tenerse en cuenta que este estudio está incompleto: no caracteriza completamente la biodistribución de los LNP que transportan su carga útil. El estudio se detuvo mientras las concentraciones en múltiples órganos seguían aumentando y, por lo tanto, no es posible decir cuáles habrían sido las verdaderas concentraciones máximas en estos órganos. No se realizaron ni planificaron estudios de seguimiento que esclarecieran el curso de tiempo completo de la distribución, el tiempo hasta la concentración máxima, las concentraciones máximas observadas y el tiempo hasta la eliminación. No se proporcionaron estimaciones de los márgenes de seguridad terapéutica.
En general, el programa de pruebas no clínicas parece lamentablemente incompleto. Este hecho se señaló claramente en el documento resumen de la inyección BNT162b2 de la Agencia Europea de Medicamentos (EMA). Los revisores comparten una admisión explícita [ 16 , p. 45] que no se han realizado estudios tradicionales de farmacocinética o biodistribución con la inyección candidata BNT162b2.
Adicionalmente, en la página 54, señalan que varios informes de la literatura indican que los ARN formulados por LNP pueden distribuirse de manera bastante inespecífica a varios órganos como el bazo, el corazón, los riñones, los pulmones y el cerebro. De acuerdo con esto, los resultados del estudio 185350 recientemente transmitido indican un patrón de biodistribución más amplio.
Aunque el estudio de biodistribución no se realizó de acuerdo con los estándares GLP de la industria, sus resultados sugieren fuertemente que las nanopartículas de lípidos con ARNm que codifica la proteína pico o espiga llegarán al torrente sanguíneo, circularán por todo el cuerpo y luego se acumularán en una variedad de órganos y tejidos. Si esto da como resultado una proteína pico expresada en esos órganos, estimulará la inmunidad y hará que esas mismas células sean atacadas por el sistema inmunitario.
La «reactogenicidad de la inyección» resultante podría parecerse a los síntomas clínicos observados con los síndromes autoinmunes de gravedad variable, en algunos casos lo suficientemente graves como para causar la muerte o una discapacidad permanente; esta conclusión está fuertemente respaldada por el estudio de autopsia antes mencionado [ 14 ]. Con el lanzamiento de las inyecciones a nivel mundial, estos tipos exactos de eventos adversos se han informado por miles en los sistemas de notificación de eventos adversos de las vacunas, pero aún ninguna agencia de salud pública ha establecido una conexión entre este mecanismo documentado preclínicamente y la alarmante situación actual de datos de resultados de salud.
2.5. Pfizer se saltó por completo las principales categorías de pruebas de seguridad
Aún más esclarecedor es lo que Pfizer decidió NO estudiar, es decir, todas las secciones de farmacología relacionadas con la seguridad y la caracterización de riesgos. Específicamente, el paquete de documentos no clínicos establece [ 3 ] :
- 2.4.2.2. Farmacodinámica secundaria No se realizaron estudios de farmacodinámica secundaria con BNT162b2.
- 2.4.2.3. Farmacología de seguridad No se realizaron estudios de farmacología de seguridad con BNT162b2 ya que no se consideran necesarios para el desarrollo de vacunas según la guía de la OMS (OMS, 2005).
- 2.4.2.4. Interacciones Farmacodinámicas de Medicamentos No se realizaron estudios no clínicos que evaluaran las interacciones farmacodinámicas de los medicamentos con BNT162b2, ya que generalmente no se consideran necesarios para respaldar el desarrollo y la autorización de productos de vacunas para enfermedades infecciosas (OMS, 2005).
- 2.4.4.4. GenotoxicidadNo se planean estudios de genotoxicidad para BNT162b2 ya que los componentes de la construcción de la vacuna son lípidos y ARN y no se espera que tengan potencial genotóxico (OMS, 2005).
- 2.4.4.5. Carcinogenicidad No se han realizado estudios de carcinogenicidad con BNT162b2 ya que los componentes de la construcción de la vacuna son lípidos y ARN y no se espera que tengan potencial carcinogénico o tumorigénico. Las pruebas de carcinogenicidad generalmente no se consideran necesarias para respaldar el desarrollo y la autorización de productos de vacunas para enfermedades infecciosas (OMS, 2005).
Revisemos qué estudios de seguridad Pfizer decidió omitir por completo.
2.5.1. ¿Qué es la Farmacología de Seguridad?
El objetivo de la farmacología de seguridad es caracterizar los aspectos farmacocinéticos/farmacodinámicos (PK/PD) de los efectos adversos de un fármaco.
- La farmacodinámica tiene como objetivo describir cómo actúa la droga en el cuerpo, mientras que
- la farmacocinética examina a qué parte del cuerpo va la droga, cuánto tiempo permanece allí y cómo se elimina.
Una ‘batería central’ de farmacología de seguridad comprende estudios para determinar los posibles efectos indeseables de un fármaco en los sistemas nervioso central, cardiovascular y respiratorio. También incluye pruebas complementarias para evaluar otros sistemas de órganos (hígado, riñón, sangre, etc.) si existen riesgos potenciales de daño a estos sistemas.
2.5.2. ¿Qué son los Estudios de Farmacodinámica Secundaria?
Las evaluaciones de nuevos fármacos para actividades farmacológicas sobre dianas distintas a la deseada terapéuticamente se denominan Farmacodinámica Secundaria. Para una clase completamente nueva de compuesto biológico con excipientes completamente nuevos y no revelados, * es completamente inaceptable omitir la evaluación de la farmacodinámica secundaria del producto completo o de sus componentes nuevos.
*Los excipientes son ingredientes formulados en el producto farmacéutico final con fines de entrega, estabilización o razones de fabricación.
2.5.3. ¿Qué son los Estudios de Interacción de Medicamentos?
Los estudios de interacciones farmacológicas están diseñados para evaluar los posibles efectos nocivos de las interacciones del nuevo producto farmacéutico con los medicamentos existentes que puede estar tomando un paciente. Por ejemplo, dicha aplicación simultánea de fármacos supondrá una carga adicional para los principales órganos metabolizadores de fármacos, en particular el hígado.
2.5.4. ¿Qué son los Estudios de Genotoxicidad y Carcinogenicidad?
Estos estudios están diseñados para evaluar el riesgo de posibles daños al ADN y los procesos celulares relacionados, y para evaluar el riesgo de promover el daño celular y la formación de cáncer. Las nuevas tecnologías basadas en ADN/ARN tienen un potencial genotóxico evidente.
Un mecanismo sencillo de daño genético es la incorporación del ácido nucleico inyectado en el genoma de la célula huésped. Esto es bien conocido con vectores virales como los derivados de adenovirus utilizados por las inyecciones de Johnson & Johnson y AstraZeneca [ 17 ] , pero también se ha demostrado recientemente con la inyección de ARNm de Pfizer en una línea celular de hígado humano [ 18 ] . Es probable que el mecanismo sea el mismo que se demostró anteriormente en el caso de la inserción genómica de secuencias derivadas del virus SARS-CoV-2, es decir, la transcripción inversa del ARNm en una copia de ADN por retrotransposón celular, seguida de la inserción cromosómica de ese copia de ADN [ 19 ]. Una segunda vía potencial hacia la genotoxicidad es la actividad metabólica de los lípidos catiónicos, que interrumpen la respiración mitocondrial y, por lo tanto, provocan la producción de especies reactivas de oxígeno, que pueden dañar químicamente el ADN [ 20 – 22 ] .
La vacuna de ARNm de Pfizer nunca se probó para excluir estos riesgos, ni en animales ni en humanos. La siguiente pregunta lógica es: ¿qué justificación se utilizó para renunciar a toda esta categoría de pruebas de seguridad farmacológica?
Cabe mencionar que Moderna realizó algunos estudios preliminares de genotoxicidad en su propia inyección de ARNm, que es muy similar a la de Pfizer. Estos estudios emplearon el ensayo de micronúcleos de eritrocitos. El llamado micronúcleo es un fragmento cromosómico que se produjo por daño cromosómico [ 23 , 24 ] y luego quedó en el citoplasma cuando se expulsó el núcleo principal. La prueba, que cuenta el número de glóbulos rojos con tales micronúcleos, se usa ampliamente para evaluar la genotoxicidad in vivo [ 24 ] . El informe de evaluación de la EMA resume y comenta lo siguiente [ 25 ] :Se realizó otro estudio de micronúcleos in vivo compatible con GLP en ratas con mRNA-1706 en nanopartículas de lípidos que contienen SM-102 mediante administración IV. En este estudio se informaron aumentos estadísticamente significativos de eritrocitos micronucleados en ambos sexos. … En los estudios toxicológicos realizados en rata, se observaron diversos efectos no genotóxicos que podrían incidir en el aumento de eritrocitos micronucleados en esta especie: hipertermia, alteración de la eritropoyesis… e inflamación del bazo, lo que podría afectar el aclaramiento de células micronucleadas del sangre.
En otras palabras, la señal positiva de genotoxicidad se atribuyó especulativamente a otras causas posibles, sin llevar a cabo ningún estudio experimental de seguimiento para decidir esta cuestión crucial.
2.6. Pfizer utilizó una interpretación deshonesta y egoísta de las pautas regulatorias para evitar las pruebas de seguridad de rutina.
Se habían documentado numerosos mecanismos de daño a los principales sistemas de órganos para la proteína pico o espiga del SARS-CoV-2 antes del lanzamiento de la inyección [ 11 , 26 – 28 ] , y los esfuerzos anteriores para desarrollar vacunas contra el virus del SARS original habían fracasado debido al aumento o mejora dependiente de anticuerpos, ADE, que hizo que a los animales vacunados les fuera peor que a los controles no vacunados tras el desafío viral [ 29 , 30 ]. Por lo tanto, había abundantes motivos de preocupación con respecto a la seguridad de la inyección basada en ARNm de Pfizer, que también desplegaría la proteína pico (spike) como antígeno. Sin embargo, Pfizer afirmó ante la FDA que los estudios de farmacología de seguridad, farmacodinámica secundaria, genotoxicidad o carcinogenicidad no eran necesarios para su producto, y como justificación de esta afirmación, Pfizer citó las Pautas para el desarrollo de vacunas de la Organización Mundial de la Salud de 2005.
El producto de Pfizer solo se reclasificó arbitrariamente como vacuna en 2020. Antes de eso, se habría clasificado como terapia génica; por lo tanto, en 2005, cuando se escribieron las pautas de la OMS, no se habría considerado una vacuna. Además, las recomendaciones de 2005 de la OMS no previeron el uso de plataformas de terapia génica para vacunas. Además, es responsabilidad de la FDA y otros organismos reguladores de todo el mundo regular la autorización y licencia de productos médicos. La OMS no tiene esta autoridad, ya que es solo un organismo no gubernamental de asesoramiento y coordinación.
2.6.1. ¿Qué afirman realmente las recomendaciones de la OMS de 2005?
Las Directrices de la OMS sobre la evaluación no clínica de vacunas [ 31 ] establecen que normalmente no se necesitan estudios de farmacocinética, pero se deben considerar caso por caso (§4.2.6), y se deben realizar estudios de toxicidad siempre que se utilicen nuevos excipientes. (y conservantes), para los que no existen datos toxicológicos (§5.2). La farmacología de seguridad está cubierta en §4.2.4, que establece: El propósito de la farmacología de seguridad es investigar los efectos de la vacuna candidata en las funciones vitales. Si los datos de estudios no clínicos y/o clínicos en humanos sugieren que la vacuna… puede afectar funciones fisiológicas (p. ej., sistema nervioso central, funciones respiratorias, cardiovasculares y renales) distintas a las del sistema inmunitario, se deben incorporar estudios farmacológicos de seguridad en la evaluación de toxicidad. Se puede encontrar información útil sobre este tema en la Nota de orientación sobre estudios de farmacología de seguridad para productos farmacéuticos humanos.
Por lo tanto, está claro que las pautas de la OMS indican la necesidad de estudios de farmacología de seguridad en casos como la proteína pico o espiga del SARS-CoV-2, con sus muchos efectos adversos documentados.
2.7. Tanto la FDA como Pfizer conocían las principales toxicidades asociadas con la clase de medicamentos de terapia génica.
Está claro que tanto el fabricante como el regulador entendieron los peligros potenciales de las vacunas basadas en genes y, por lo tanto, no pueden alegar falta de conocimiento anticipado de estos riesgos. Esto apunta a fraude intencional y colusión entre Pfizer y los reguladores para impulsar este producto peligroso no probado en el mercado.
Existen varios documentos de orientación de la FDA para el estudio de productos de terapia génica y celular en investigación, incluidas las vacunas terapéuticas [ 2 , 32 – 35 ] . Estos documentos de orientación contienen pensamiento regulatorio que anticipa claramente muchos riesgos con esta clase de producto. Específicamente, el documento de orientación sobre estudios no clínicos de 2013 [ 32 ] establece que: Se recomienda encarecidamente el uso de estudios in vitro para la identificación de posibles problemas de seguridad y MOA [mecanismo de acción] de un producto CGT [terapia celular y génica] en investigación. Sin embargo, esta prueba por sí sola no es suficiente para anticipar de forma fiable el resultado de la integración fisiológica y funcional del producto tras la administración in vivo. En consecuencia, el programa de pruebas preclínicas debe incorporar un enfoque multifactorial gradual para lograr una comprensión de la plausibilidad biológica para el uso del producto CGT (terapia celular y génica) en investigación en la población de pacientes prevista. Para las pruebas preclínicas in vivo, se recomienda el uso de modelos animales de enfermedad/lesión, ya que tales estudios permiten la caracterización de los cambios morfológicos resultantes junto con cambios funcionales/conductuales observables.
El documento de orientación de la FDA sobre el programa de ensayos clínicos de fase temprana de 2015 [ 2 ] es extenso y advierte sobre los riesgos graves conocidos de la experiencia previa con terapias génicas:
- insuficiencia multiorgánica y muerte,
- inducción de tumores y cánceres,
- leucemia de células T de inicio tardío,
- expresión génica incontrolablemente prolongada incluso después de una sola administración,
- autoinmunidad,
- expresión alterada de los genes de la célula huésped,
- migración del producto a sistemas de órganos no deseados, y
- desprendimiento de partículas virales transgénicas que podrían transmitirse a otros individuos.
La guía también establece que los riesgos asociados con la clase de terapia génica pueden ser completamente nuevos y no pueden derivarse del historial previo de otras clases de medicamentos. En otras palabras, esta clase es excepcionalmente arriesgada y requiere un extenso y riguroso programa de pruebas de seguridad. Debido a estas toxicidades potencialmente graves, las pautas de la FDA generalmente recomiendan no realizar tales estudios en voluntarios sanos.
Cabe señalar que antes de 2020, todos los productos derivados de la terapia génica se estaban desarrollando para enfermedades extremadamente graves, a menudo mortales, como el cáncer terminal y la enfermedad de Huntington. Dichos medicamentos ni siquiera podrían probarse en personas sanas, mucho menos prescribirse a todos los humanos del planeta como tratamiento profiláctico, y mucho menos forzarse a todos los seres humanos independientemente del consentimiento.
El documento no clínico de Pfizer establece que la compañía consideró las pautas de la FDA sobre el desarrollo de vacunas contra el COVID-19 [ 33 ] . No está claro, sin embargo, que se haya realizado alguna consideración de este documento, ya que ninguna de sus recomendaciones se implementó en las evaluaciones no clínicas de Pfizer. También debemos preguntarnos por qué se utilizaron las recomendaciones de la OMS de 2005, en lugar del documento de orientación de la industria de la FDA de 2020, como base para el diseño del programa de pruebas no clínicas. Específicamente, la guía de la FDA establece claramente que: Para una inyección candidata contra el COVID-19 que consiste en un tipo de producto novedoso y para el cual no hay datos clínicos y no clínicos previos disponibles, se requerirán estudios de seguridad no clínicos antes de proceder a los ensayos clínicos FIH 21 CFR 312.23(a)(8).
La guía de la FDA expresa específicamente su preocupación por el aumento de la enfermedad respiratoria asociada a la inyección y la necesidad de caracterizar y excluir este riesgo con el nuevo producto de la inyección: Los datos de estudios en modelos animales a los que se les administró ciertas construcciones de vacunas contra otros coronavirus (SARS-CoV y MERS-CoV) han generado preocupaciones sobre un riesgo teórico de enfermedad respiratoria mejorada o aumentada (ADE) asociada a la vacuna COVID-19. En estos estudios, a los modelos animales se les administraron construcciones de vacunas contra otros coronavirus y, posteriormente, se desafiaron con el virus de tipo salvaje respectivo. Estos estudios han mostrado evidencia de reacciones pulmonares inmunopatológicas características de una hipersensibilidad de tipo Th-2 similar a la ERD descrita en bebés y animales a los que se les administró la vacuna del virus sincitial respiratorio (RSV) inactivado con formalina y que posteriormente fueron desafiados con el virus RSV debido a la exposición natural. o en el laboratorio, respectivamente (Refs. 4-9). Los candidatos a vacunas deben evaluarse a la luz de estos estudios.
Después de 2000, los científicos hicieron muchos intentos para crear inyecciones contra el coronavirus. Durante los últimos 20 años, todo terminó en fracaso porque los animales en los ensayos clínicos se enfermaron gravemente y muchos murieron, al igual que los niños en la década de 1960. Puedes leer un resumen de esta historia / ciencia aquí . O si desea leer los estudios individuales, puede consultar estos enlaces:
- En 2004, el intento de vacunación produjo hepatitis en hurones.
- En 2005, los ratones y las civetas se enfermaron y se volvieron más susceptibles a los coronavirus después de ser vacunados.
- En 2012, los hurones enfermaron y murieron. Y en este estudio , los ratones y hurones desarrollaron una enfermedad pulmonar.
- En 2016, este estudio también produjo enfermedad pulmonar en ratones.
Dado este potencial conocido para la mejora o aumento de la enfermedad dependiente de anticuerpos (ADE), es aún más desconcertante que Pfizer decidiera ignorar estas pautas y que la FDA decidiera dejar que se salieran con la suya.
Hay un párrafo del documento de orientación que describe las condiciones bajo las cuales se podrían renunciar a los estudios de seguridad no clínicos: En algunos casos, puede que no sea necesario realizar estudios de seguridad no clínicos antes de los ensayos clínicos de FIH porque la información adecuada para caracterizar la seguridad del producto puede estar disponible de otras fuentes. Por ejemplo, si la inyección candidata contra la COVID-19 se elabora utilizando una tecnología de plataforma utilizada para fabricar una inyección con licencia u otras inyecciones en investigación previamente estudiadas y está suficientemente caracterizada, es posible que se puedan usar datos toxicológicos (p. ej., datos de estudios de toxicidad de dosis repetidas , estudios de biodistribución) y datos clínicos acumulados con otros productos que utilizan la misma plataforma para respaldar los ensayos clínicos de la FIH para esa vacuna candidata contra el COVID-19. Los fabricantes de vacunas deben resumir los hallazgos y proporcionar una justificación si consideran usar estos datos en lugar de realizar estudios de seguridad no clínicos.
Es cuestionable si este párrafo se inserta para proporcionar a Pfizer y Moderna un futuro «fuera» de las pruebas de seguridad al afirmar que sus productos son derivados de «productos seguros aprobados» mientras se levanta una barrera regulatoria para otros fabricantes que desearían diseñar una vacuna diferente contra el Covid-19?
En general, la interpretación deshonesta de las pautas por parte de Pfizer y la selección selectiva de las reglamentaciones aplicables dieron como resultado un descarado desprecio por todas las evaluaciones de seguridad de rutina. Es inaceptable que un fabricante farmacéutico no estudie el potencial de su producto para dañar los principales sistemas de órganos, o que sustituya el producto con un sustituto o una versión diferente, afirme la comparabilidad teórica y luego afirme que no hay riesgos para los principales sistemas de órganos humanos. . ¡La ausencia de evidencia de daño no es evidencia de ausencia de daño!
El mandato de la FDA como regulador de la industria requiere que la agencia cuestione y verifique tal desprecio imprudente por las pruebas de seguridad. Un regulador honesto habría cuestionado la afirmación del fabricante de que las principales categorías de estudios de seguridad no eran aplicables a su producto. Esto no se puede explicar por incompetencia. La FDA cuenta con profesionales calificados y experimentados en farmacología y toxicología. En este punto, con millones de informes de eventos adversos que se acumulan rápidamente en todas las bases de datos de salud pública, ni la FDA, NIH, CDC, Pfizer ni otros fabricantes pueden afirmar que ignoran estos problemas. Debe plantearse la cuestión del fraude y la negligencia deliberada tanto de los fabricantes como de los reguladores.
Referencias
- Anonymous, (2022) Guidance, Compliance & Regulatory Information (Biologics).
- Anonymous, (2015) Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products: Guidance for Industry.
- Anonymous, (2022) FDA records pertaining to non-clinical studies on the Pfizer COVID-19 vaccine, released pursuant to FOIA request 2021-4389.
- Anonymous, (2022) Principles of Premarket Pathways for Combination Products: Guidance for Industry and FDA Staff.
- Fink, D. (2020) FDA letter to BioNTech, granting fast track designation for their BNT162b1 and BNT162b2 vaccines.
- BioNTech, (2020) Investigator’s Brochure BNT162/PF-07302048.
- Mulligan, M.J. et al. (2020) Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586:589-593
- FDA, (2021) Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial.
- Government, U. (2022) FDA regulation: Good Laboratory Practice for Nonclinical Laboratory Studies.
- Patel, S. et al. (2020) Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 11:983
- Buzhdygan, T.P. et al. (2020) The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol Dis 146:105131
- Raghavan, S. et al. (2021) SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Frontiers Cardiovasc Med 8 (preprint)
- Kakarla, V. et al. (2021) Pathophysiologic mechanisms of cerebral endotheliopathy and stroke due to Sars-CoV-2. J. Cereb. Blood Flow Metab. 41:1179-1192
- Bhakdi, S. and Burkhardt, A. (2021) On COVID vaccines: why they cannot work, and irrefutable evidence of their causative role in deaths after vaccination.
- Palmer, M. and Bhakdi, S. (2021) The Pfizer mRNA vaccine: Pharmacokinetics and Toxicity.
- Anonymous, (2021) EMA Assessment report: Comirnaty.
- Doerfler, W. (2021) Adenoviral Vector DNA- and SARS-CoV-2 mRNA-Based Covid-19 Vaccines: Possible Integration into the Human Genome—Are Adenoviral Genes Expressed in Vector-based Vaccines?. Virus Res. 302:198466
- Alden, M. et al. (2022) Intracellular Reverse Transcription of Pfizer BioNTech COVID-19 mRNA Vaccine BNT162b2 In Vitro in Human Liver Cell Line. Current issues in molecular biology 44:1115-1126
- Zhang, L. et al. (2021) Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. U. S. A. 118 (preprint)
- Knudsen, K.B. et al. (2015) In vivo toxicity of cationic micelles and liposomes. Nanomedicine 11:467-77
- Lv, H. et al. (2006) Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 114:100-9
- Yun, C. et al. (2016) Cargo-Free Nanoparticles Containing Cationic Lipids Induce Reactive Oxygen Species and Cell Death in HepG2 Cells. Biol. Pharm. Bull. 39:1338-46
- Heddle, J.A. et al. (1983) The induction of micronuclei as a measure of genotoxicity. A report of the U.S. Environmental Protection Agency Gene-Tox Program. Mutat. Res. 123:61-118
- Sommer, S. et al. (2020) Micronucleus Assay: The State of Art, and Future Directions. Int. J. Mol. Sci. 21 (preprint)
- Anonymous, (2021) EMA Assessment report: COVID-19 Vaccine Moderna.
- Seneff, S. and Nigh, G. (0) Worse Than the Disease? Reviewing Some Possible Unintended Consequences of the mRNA Vaccines Against COVID-19. 2:38-79
- Suzuki, Y.J. et al. (2021) SARS-CoV-2 spike protein-mediated cell signaling in lung vascular cells. Vascul. Pharmacol. 137:106823
- Lee, W.S. et al. (2020) Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 5:1185-1191
- Du, L. et al. (2009) The spike protein of SARS-CoV–a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 7:226-36
- Tseng, C. et al. (2012) Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS One 7:e35421
- Anonymous, (2005) WHO guidelines on non-clinical evaluation of vaccines, Annex 1, TRS No 927.
- Anonymous, (2013) Preclinical Assessment of Investigational Cellular and Gene Therapy Products.
- Anonymous, (2020) Development and Licensure of Vaccines to Prevent COVID-19.
- Anonymous, (2021) Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial.
- Anonymous, (2022) Human Gene Therapy Products Incorporating Human Genome Editing.



